







The progress that has been achieved in the field of pulmonary arterial hypertension (PAH) in the past two decades is one of the most remarkable advances accomplished. Intense research focusing on the cellular, molecular, and genetic basis of pulmonary vascular diseases has enhanced our understanding of this devastating illness, through which a systematic approach for diagnosis and approval of eight PAH- directed treatments has been developed. As a result, for a disease that was until recently considered untreatable and uniformly fatal, significant improvements in quality of life and survival have been achieved. This article will focus on current concepts in mechanisms, diagnosis, and therapeutic approaches for PAH.
Classification
Pulmonary hypertension (PH) was originally grouped as either primary pulmonary hypertension (PPH) or secondary pulmonary hypertension. With enhanced understanding of the disease mechanism and the recognition that certain types of PH should be classified together based on shared pathology and responses to treatment, focus has been directed to unifying the classification of different types of PH. The most recent classification, from the 4th World Symposium at Dana Point, instituted several modifications to the previous Venice Classification (see Table 1).1 Specifically, the term ‘heritable PAH’ has replaced ‘familial PAH’ since studies have shown that the main genetic abnormality discovered to date, bone morphogenic protein receptor type 2 (BMPR2) mutation, has been detected in 11–40% of idiopathic PAH (IPAH; a term that has replaced PPH) patients with no family history. Thus, all patients with a BMPR2 mutation are grouped as heritable PAH whether the patient is identified in conjunction with other family members or diagnosed with a de novo mutation.1–3 Another major modification is moving PAH associated with schistosomiasis from the thromboembolic PH (Group 4) to a subgroup of PAH (Group 1). Schistosomiasis affects over 200 million people worldwide. Recent studies have shown that PH associated with schistosomiasis manifests similar pathological changes and clinical presentations to IPAH.4,5 Another noteworthy modification is adding methamphetamine as a ‘very likely’ risk factor for developing PAH.1
Epidemiology
The PPH Registry, which collected epidemiological and outcomes data from 32 centers on 187 patients with IPAH from 1981 to 1985, showed PPH to be a rare disease, with an estimated annual incidence of two to five per million persons in the US. The mean age at diagnosis was 37 years, and the female:male ratio was 1.7:1.6 More recent registries show that the prevalence of PAH is approximately 15–20 cases per million population.7 Furthermore, the age of patients being diagnosed with PAH is higher, with patients in their 70s being reported, and female preponderance greater—findings that are being corroborated by data from the Registry to Evaluate Early and Long Term PAH Disease Management (REVEAL).8,9 Among all the associated conditions, patients with connective tissue disease (CTD) are at greatest risk for developing PAH. For patients with systemic sclerosis, echocardiographic-based screening has found a prevalence of PAH of between 7 and 12%.10,11 PAH associated with systemic sclerosis has worse outcome compared with IPAH patients, even with current treatments.12,13 Clinical studies are ongoing evaluating the most effective methods of early detection of PAH in CTD patients.

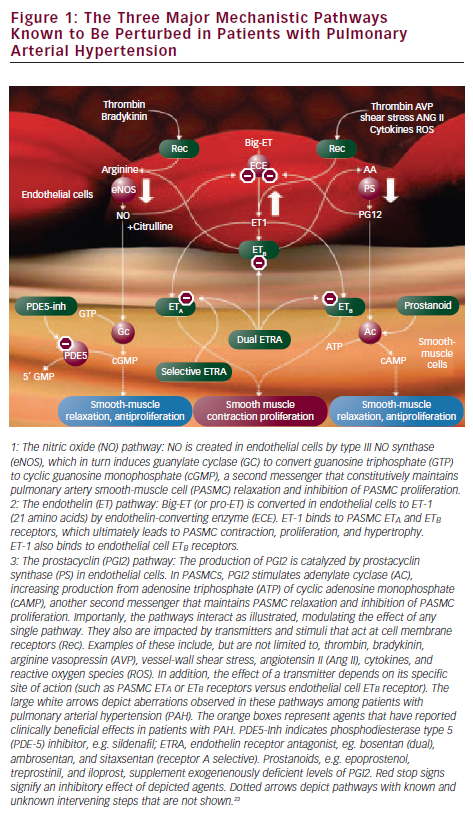
Pathobiology
Pulmonary arterial hypertension is defined as chronic elevation of mean pulmonary arterial pressure (mPAP) >25mmHg and pulmonary capillary wedge pressure (PCWP) ≤15mmHg.14 It is characterized by a progressive increase in pulmonary vascular resistance (PVR), leading to right ventricular (RV) dysfunction, RV failure, and death. The increase in PVR is caused by loss of vascular luminal cross-sectional area due to pulmonary vascular remodeling, which occurs as a consequence of excessive cellular proliferation and reduced apoptosis due to multiple pathological abnormalities, including endothelial dysfunction, inflammatory and growth factor dysregulation, and chronic vasoconstriction. The histological changes in PAH involve all layers of the vessel wall and include intimal hyperplasia, medial hypertrophy, adventitial proliferation, and in situ thrombosis.15,16
The process involved in disease initiation and propagation has been described as a ‘multiple-hit hypothesis,’ a concept referring to the interaction of an inciting stimulus on an individual with a predisposing state.16,17 Two or more ‘hits’ are thought to consist of a genetic abnormality or substrate that renders an individual susceptible—such as a systemic disorder, an environmental trigger, or additional genetic mutations—which can activate systemic and endothelial changes leading to vasoconstriction, cellular proliferation, and prothrombotic state.
Perhaps the greatest contribution towards formulating PAH-directed therapies is derived from enhanced knowledge of the pulmonary vascular endothelium, which, in a normal state, tightly regulates the production of vasodilators and vasoconstrictors to maintain a low- pressure system. In PAH, an imbalance of mediators occurs with decreased production of the mediators that produce vasodilation and control smooth-muscle cell proliferation and platelet aggregation (prostacyclin and nitric oxide [NO]), and increased synthesis of mediators that promote vasoconstriction, cellular proliferation, and platelet aggregation (endothelin-1 [ET-1]). Prostacyclin, produced by both endothelial and smooth-muscle cells in the vasculature, is deficient in PAH along with its key enzyme, prostacyclin synthase.18 The effects of NO are mediated through its second messenger, cyclic guanosine monophosphate (cGMP), which is degraded by phosphodiesterase (PDE). PDE-5 is abundant in the pulmonary vasculature and its inhibitor promotes the accumulation of intracellular cGMP, thereby enhancing NO-mediated effects.19 The plasma level of ET-1 is increased in PAH and its level has been shown to be inversely proportional to the magnitude of the pulmonary blood flow and cardiac output.20 ET-1 acts via two receptors, ETA and ETB.21 Activation of the ETA and ETB receptors located on smooth-muscle cells induces vasoconstriction and cellular proliferation and hypertrophy. By contrast, stimulation of ETB receptors on endothelial cells results in production of vasodilators and is involved in the clearance of ET-1 from the circulatory system22,23 (see Figure 1).
Diagnosis
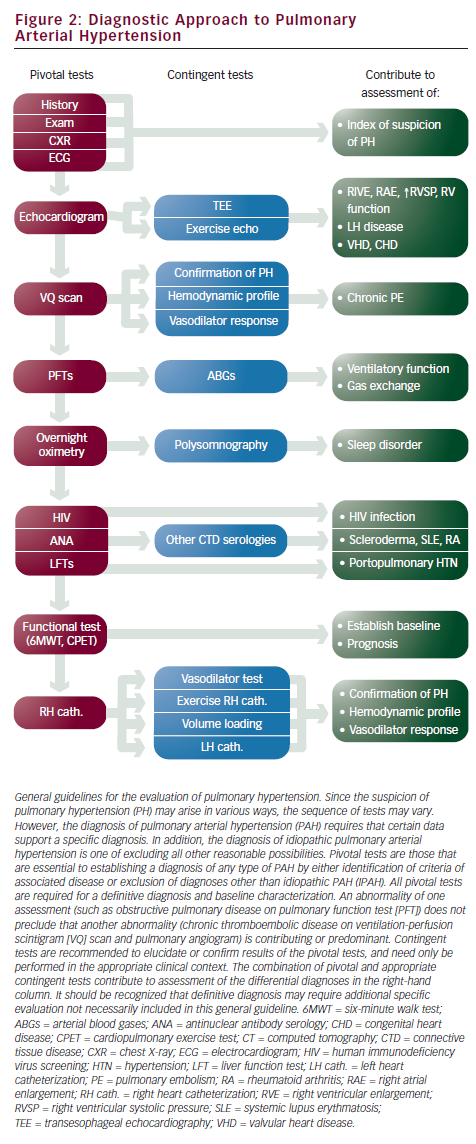
PAH poses a diagnostic challenge due to the relative rarity of the condition, the non-specific nature of the presenting symptoms, and the inability to detect the disease using routine outpatient tests. However, certain clinical features can serve as indicators to raise suspicion. The most common presenting symptoms are exertional dyspnea and fatigue.6,14,24 However, patients presenting with pre-syncope or syncope without obvious cause, especially in a young female, should raise the possibility of PAH.23–25 Recognizing clinical risk factors for PAH can also assist in earlier diagnosis; these include patients with BMPR2 mutation, first-degree relatives of patients with BMPR2 mutation, prior appetite suppressant use, or patients with the associated conditions recognized for increased incidence of PAH (systemic sclerosis, HIV infection, portal hypertension, congenital heart disease with shunt, sickle cell disease, recent acute pulmonary embolism).24
Transthoracic Doppler echocardiography is the best method to screen patients suspected of PAH. It provides an estimation of pulmonary artery systolic pressure using the modified Bernoulli equation 4v2, in which v is the velocity of the tricuspid regurgitation (TR) jet in meters per second. The right ventricular systolic pressure (RVSP) is derived by adding the estimated value of right atrial pressure (RAP) to the pressure gradient (RVSP = 4v2 + RAP). The accuracy of RVSP estimation can be influenced by the quality of the TR jet envelope, the patient’s body habitus, the accuracy of RAP estimation used, and operator competency.26–28 The recommendation from the 4th World Symposium states that a reasonable approach may be to consider TR velocity >2.8m/second and tricuspid insufficient peak gradient ≥31mmHg at rest as elevated, except in elderly and/or very obese patients.14,29 Echocardiographic findings indicative of advanced disease and poor prognosis in PAH are RV dilatation and dysfunction, right atrial enlargement, presence of pericardial effusion, and septal displacements.30
Echocardiogram is necessary to evaluate for other cardiac conditions that can elevate pulmonary artery pressures (PAPs), namely congenital heart disease, heart failure (systolic and diastolic), and valvular heart disease. Evaluating for the presence of diastolic dysfunction is one of the more challenging aspects of PAH evaluation for several reasons: it is the most common diagnosis that is mistakenly interpreted as PAH; diastolic dysfunction and diastolic heart failure are common disorders among older patients with risk factors (i.e. systemic hypertension, diabetes, coronary artery disease, atrial fibrillation); and treatment for PAH and diastolic dysfunction are distinctly different and misdiagnosis can lead to harm, since some PAH-specific therapies can worsen pulmonary venous hypertension and precipitate pulmonary edema.31,32 A finding of left atrial enlargement, even in the absence of obvious left ventricular (LV) dysfunction, should raise the suspicion of elevated left- sided pressures and pulmonary venous hypertension rather than PAH. In these patients, it is critical to obtain a reliable PCWP during right heart catheterization; if a satisfactory PCWP wave form cannot be obtained, left heart catheterization may need to be performed to accurately measure the LV end-diastolic pressure.
Assessing for the presence of associated conditions that can contribute to PAH or cause PH requires checking for serologies for CTD, hepatic disease, and HIV. A pulmonary function test is needed to rule out significant pulmonary disorder as a causative factor. A complete evaluation for chronic thromboembolic pulmonary hypertension (CTEPH) is critical to determine whether a patient is a candidate for thromboendarectomy, a surgical intervention that can reverse pulmonary hypertension and potentially render a cure for this disease.33 Screening with ventilation perfusion (VQ) scan is recommended, followed by pulmonary angiography if abnormal (see Figure 2).
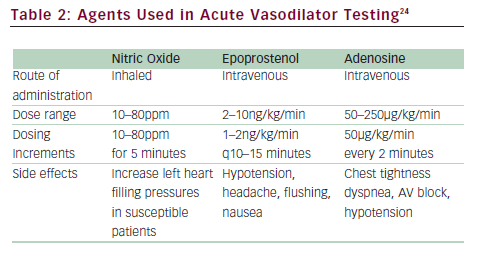
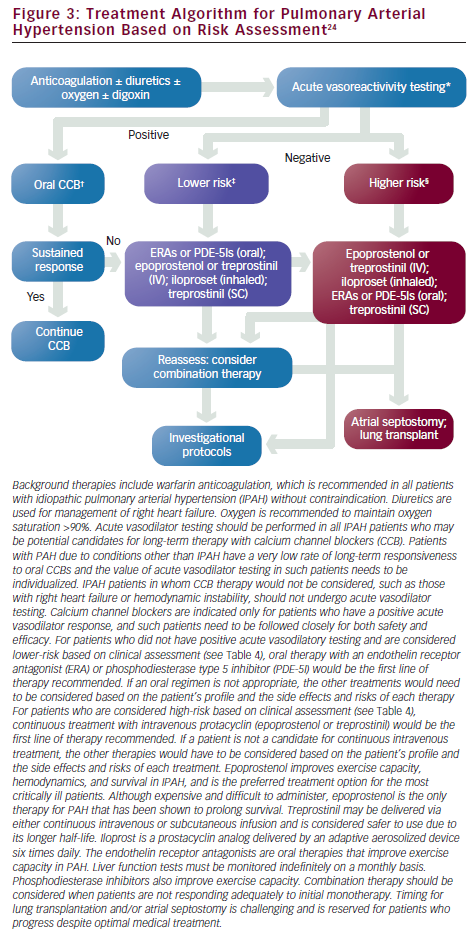
Right heart catheterization is necessary to diagnose PAH. Hemodynamic measurements are needed to confirm the elevation of pulmonary artery pressures (mPAP >25mmHg) and to exclude left-sided heart failure (PCWP ≤15mmHg). Elevation of PVR >3 Wood units has been included in some definitions of PAH. Hemodynamic parameters also provide critical prognostic information since elevated RAP and decreased cardiac index are markers of poor prognosis.34,35 Acute vasodilator challenge is recommended for IPAH patients at the time of initial diagnostic right heart catheterization to identify those who demonstrate significant decrease in pulmonary artery pressures to determine whether high-dose calcium channel blocker (CCB) therapy may be appropriate. The current definition of a responder is a reduction of mPAP by at least 10mmHg to a value of 40mmHg or less with stable cardiac output (see ‘Conventional Therapies’).24,36 The agent of choice for acute vasodilatory testing is NO, a short-acting selective pulmonary vasodilator that is administered via a face mask during right heart catheterization. It is well tolerated with very few systemic effects, although it is costly. Intravenous prostacyclin and adenosine have also been used37–39 (see Table 2).
Approved Therapies
Currently, eight therapies have been approved by the US Food and Drug Administration (FDA) for PAH, specifically aimed at augmenting prostacyclin deficiency (epoprostinil, treprostinil, iloprost), increasing the effect of NO via PDE-5 inhibitors (sildenafil, tadalafil), and blocking the actions of ET-1 with endothelin receptor antagonists (ERAs; bosentan, ambrosentan) (see Table 3).
Prostacyclins
Epoprostenol is the first therapy to be approved by the FDA for IPAH and is considered the ‘gold standard’ among treatments. It remains the only PAH drug to demonstrate survival benefit during a clinical trial. The landmark study conducted on 81 patients with functional class III or IV IPAH comparing epoprostenol and conventional therapy versus conventional therapy alone demonstrated improvement in six-minute walk distance (6MWD), hemodynamics, and quality of life with epoprostenol treatment.40 There were eight patients who died during the 12-week study, all randomized to conventional therapy (p=0.003). The benefits of long-term treatment with epoprostenol in IPAH patients were reported from retrospective analysis from two large centers showing improvements in functional class, 6MWD, hemodynamics, and survival (compared with historical or calculated survival data).35,41 Epoprostenol has also been studied in PAH associated with the scleroderma spectrum of diseases, HIV, congenital heart disease, and portopulmonary hypertension.42–45
There are several challenges to using epoprostenol. Due to its short half-life (less than six minutes), it needs to be administered by continuous intravenous infusion via a tunneled catheter. Any interruption of the infusion can be potentially life-threatening. Careful patient selection with appropriate family support is critical since the patient must be able to properly mix the medication, care for the catheter, and manage the pump. The greatest risk involves catheter- related infections and malfunction, which can cause significant morbidity (0.1–0.6 cases per patient-year).41 The drug is started in a closely monitored inpatient setting at 2ng/kg/minute and is titrated following efficacy and side effects, which include headache, flushing, diarrhea, and nausea—all common side effects seen with prostanoids. Dosing needs to be highly individualized, but most experts agree that for the majority of patients the optimal dose for chronic therapy is between 25 and 40ng/kg/minute. Chronic overdose can lead to high output failure, so periodic right heart catheterization is needed to ensure appropriate dosing.24,46 Given the enormous complexity, it is recommended that patients requiring epoprostenol be referred to experienced PH centers.
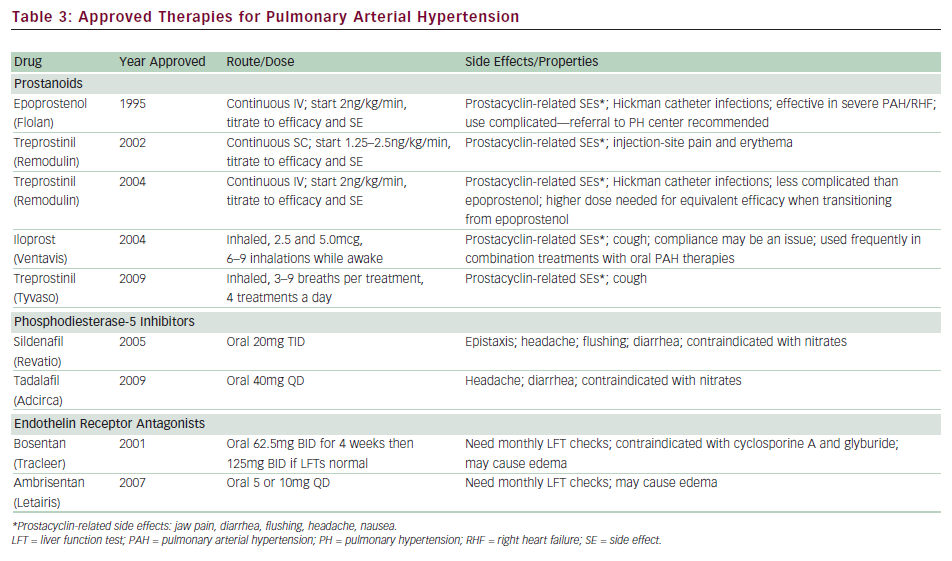
Treprostinil is a stable prostacyclin analog with a half-life of four to five hours that is currently available for administration via subcutaneous, intravenous, and inhaled routes. Treprostinil was first developed for subcutaneous infusion and received approval after demonstrating a modest increase in 6MWD during a 12-week placebo-controlled, randomized trial of 470 patients.47 The difficulty in using the subcutaneous delivery system stems from pain and erythema at the site of the infusion, which deterred dose escalation during the clinical trial period. Long-term experience has shown that the site pain is not dose- related, and that most patients experience less discomfort after appropriate dose titration to alleviate their PAH symptoms. Long-term observational reports have recently shown improvements in exercise capacity, with survival benefits.48
Due to limitations posed by site pain, treprostinil was studied using an intravenous route in a 12-week open-label trial of 16 patients.49 Although a small study, it demonstrated the safety and efficacy of intravenous infusion. This was followed by a transition study among 31 stable patients on epoprostenol who were switched to intravenous treprostinil.50 Among the 27 patients who completed the study, the 6MWD was maintained. The most noteworthy finding was that the dose of intravenous treprostinil required to maintain equivalent efficacy was about twice that of epoprostenol (83ng/kg/minute versus 40ng/kg/minute). Intravenous treprostinil was approved by the FDA in 2004 for functional class II, III, and IV PAH patients in whom subcutaneous infusion is not tolerated. Some potential advantages over epoprostenol are that it is thought to be safer due to its longer half-life from possible rebound effects in cases of infusion interruption, and that it is easier to administer. A tunneled catheter is still needed and a Centers for Disease Control and Prevention (CDC) report recently raised concerns over increased risk of bloodstream infections, especially with Gram-negative organisms.51 Although no definite conclusion could be drawn from these reports, a revision in recommendations regarding catheter care was placed in effect.52 An inhaled form of treprostinil has recently been approved for PAH. This was studied in a randomized, double-blind, placebo-controlled study of 235 IPAH or APAH patients on top of background oral treatments (Treprostinil Sodium Inhalation Used in the Management of Pulmonary Arterial Hypertension [TRIUMPH]).53 An oral formulation of treprostinil is currently undergoing phase III clinical trial.
Iloprost is a prostacyclin analog with a half-life of approximately 20 minutes, available as an inhaled form in the US. The pivotal study, conducted in Europe, evaluated 207 functional class III and IV patients in a 12-week study (Aerosolized Iloprost Randomized Study [AIR]).54 A novel composite end-point of improvement in functional class by at least one level and improvement in 6MWD by at least 10% in the absence of clinical deterioration was set as the primary end-point, which was met by 16.8% of the treated group and 4.9% of the placebo group (p=0.007). It is recommended that patients administer treatments six to nine times a day while awake. A potential advantage is the ability to deliver prostacyclin directly to the pulmonary bed, thereby minimizing systemic side effects and not requiring indwelling catheters. The need for frequent treatments has been noted as a major disadvantage. Common side effects include cough, headache, flushing, and jaw pain.
Phosphodiesterase Inhibitors
Sildenafil was studied among 278 PAH patients in a 12-week randomized, placebo-controlled trial (Sildenafil Use in Pulmonary Arterial Hypertension [SUPER]).55 Treatment resulted in significant improvements in 6MWD, which was similar among the three doses studied (45, 46, and 50m for 20, 40, and 80mg, respectively). Functional class and hemodynamics also improved; however, there was no difference in rate of clinical worsening. One-year outcome data on 222 patients demonstrated sustained improvement in 6MWD; however, nearly all patients were titrated up to 80mg TID. Some of the reported side effects included headache and epistaxis, although the drug was, overall, well tolerated. Reports regarding visual disturbances have raised some concerns, especially among diabetics and patients with cardiovascular risk factors, and some practitioners advocate obtaining baseline ophthalmological evaluations.
Tadalafil is a once-a-day PDE-5 inhibitor that was recently studied among 392 patients in a 16-week placebo-controlled study (Pulmonary Arterial Hypertension and Response to Tadalafil [PHIRST]).56 The treated groups received 2.5, 10, 20, and 40mg doses. Tadalafil increased 6MWD in a dose-dependent manner, and treatment with 40mg resulted in a statistically significant increase in 6MWD. Furthermore, the 40mg dose group demonstrated improvement in time to clinical worsening. The most frequent side effects included headache (which appears to be dose-related), myalgia, and flushing.
Endothelin Receptor Antagonists
Bosentan, a dual endothelin receptor blocker, was the first oral therapy to be approved for PAH. The pivotal study, Bosentan Randomized trial of Endothelin Antagonist THErapy-1 (BREATHE-1), was conducted in 213 patients with functional class III–IV PAH, demonstrating a significant improvement in 6MWD as well as time to clinical worsening, which was defined as death, lung transplantation, hospitalization for PH, lack of clinical improvement, or worsening leading to discontinuation, need for epoprostenol therapy, or atrial septostomy.57 Longer-term observational findings in patients treated with open-label bosentan as first-line therapy have demonstrated improved survival compared with National Institutes of Health (NIH) formula-based calculations.58 Bosentan has also been studied in patients with associated PAH conditions such as Eisenmenger’s syndrome, HIV, and inoperable CTEPH.59–61
Bosentan is metabolized primarily through the hepatic P450 enzyme systems. The reported increase in transaminases more than three times upper limit of normal (ULN) is about 10–12%, which is dose- dependent and reversible.57 All patients receiving bosentan are required to have liver function tests before drug initiation and then monthly. Hemoglobin should be checked every three months since bosentan therapy has been associated with anemia. The other side effect is edema, which may require diuretic adjustments in some patients. Bosentan has been shown to have teratogenic effects in animals, so is contraindicated in pregnancy. It may decrease the efficacy of hormonal contraception, so women of childbearing age must be counseled not to rely only on oral contraception. Glyburide and cyclosporine A are contraindicated with bosentan due to significant drug interaction and potential for liver damage. There is no significant interaction with warfarin and bosentan.
Ambrisentan is a selective ETA receptor antagonist that was studied in the pivotal trials ARIES I and II, conducted in the US and Europe/South America, respectively.62 The combined studies enrolled approximately 400 patients in a 12-week placebo-controlled study, demonstrating significant increases in 6MWD, delayed time to clinical worsening, and improvement in functional class. Ambrisentan carries the same warning as bosentan regarding potential for liver toxicity and requires monthly liver function test evaluation. However, the enzymatic routes differ compared with bosentan and the incidence of liver function test abnormalities was lower in the pivotal studies (incidence of aminotransferases >3 x ULN 0.8%).62 This was further investigated in a study of 36 patients who experienced increases in hepatic enzymes on bosentan or sitaxsentan, demonstrating that ambrisentan was well tolerated in this cohort.63 Peripheral edema, another side effect of ERAs, was found to be more clinically significant among older patients, which prompted the FDA to place a label warning.64 Similar to bosentan, ambrosentan is teratogenic and the same warnings apply regarding pregnancy.
Conventional Therapies
Treatment with calcium channel blockers (CCBs) is based on the retrospective analysis of clinical outcome of 557 IPAH patients, of whom 70 (12.6%) demonstrated acute vasoreactivity and were placed on CCBs.36 Only 6.8% of patients remained stable for more than one year on CCBs. The current recommendation states that CCBs should be considered as initial therapy for “patients with IPAH in the absence of right heart failure, demonstrating a favorable-acute response to vasodilator,” which is defined as a fall in mPAP of at least 10mmHg to ≤40mmHg, with an increased or unchanged cardiac output.24,36,65 Empirical treatment with CCBs is contraindicatied. For patients with PAH associated with other underlying processes such as scleroderma or congenital heart disease, the number of patients who benefit from CCBs long term is even lower and the net benefit of vasodilator testing is at best weak and not likely to influence choice of therapy.24,36 Close follow-up is critical to assess clinical response for those who meet the criteria. Amlodipine is the most commonly used agent along with diltiazem or long-acting nifedipine. Verapamil is best avoided due to its negative inotropic effect. If the patient does not improve to functional class I or II on CCBs, PAH-specific therapy needs to be started. Warfarin anticoagulation is recommended for all IPAH patients in the absence of contraindications, and the recommended international normalized ratio (INR) among expert consensus is 1.5–2.5.24,65,66 For patients with PAH due to scleroderma and hepatic disease, the increased risk for bleeding must be considered.
Treatment Strategies
The goals in treating patients with PAH are to improve quality of life and survival. Although outcome is better, the reality is that the majority of patients completing clinical trials failed to improve their functional class by one and approximately one-third to half the patients did not achieve a 6MWD >380m, a distance that has been shown to correlate with outcome.40,42,47,54,55,57 Furthermore, the key determinant of survival in patients with PAH is the preservation of RV function.67 This was first reported from the PPH Registry and confirmed in more recent studies in which elevated RAP and decreased cardiac index were two major factors associated with mortality.34,35
Risk Stratification in Determining Therapy
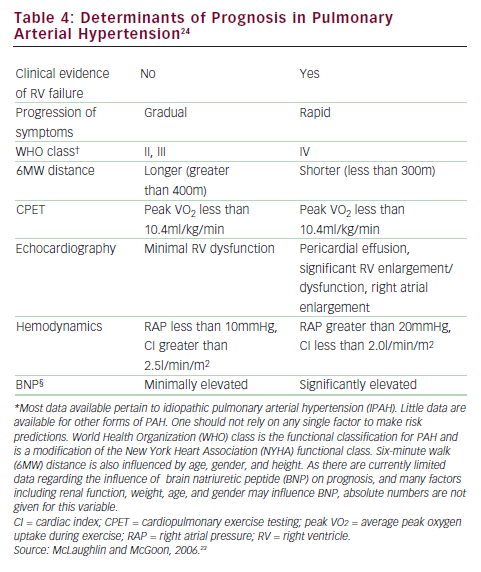
One of the most pressing challenges in PAH is to determine how to best utilize current therapies to further improve outcome. Risk stratification that incorporates several proven prognostic factors to guide choice of treatment is widely used in clinical practice (see Figure 3).23,24 In patients with low-risk profiles, initiation with an oral regimen is recommended. In those with high-risk profiles (low 6MWD, high RAP, low cardiac index), starting with more aggressive treatment is warranted (see Table 4).23,24
Early Initiation of Treatment in Mildly Symptomatic Patients
Another key concept that has emerged is treating mildly symptomatic patients based on the results of the Endothelin Antagonist tRial in miLdlY symptomatic PAH (EARLY) trial, which enrolled 168 PAH patients in a double-blind, placebo-controlled trial with bosentan.68 This was the first PAH study that exclusively enrolled functional class II patients who had distinctly higher baseline 6MWD (438±86m) than all other previous clinical trial participants (~350m). A significant decrease in PVR was seen in the treated group at the end of six months compared with the placebo group, a reliable assessment of improvement in RV function. Furthermore, a significant delay in clinical worsening was reported among the treated group, demonstrating that when left untreated, even mildly symptomatic patients progress hemodynamically and symptomatically.
Combination Therapy
The goals of utilizing combination treatment are to enhance efficacy while minimizing toxicity for patients who lack sufficient response or demonstrate worsening on monotherapy. Although many small single- center experiences have been reported, only three randomized placebo-controlled trials have been completed to date. The BREATHE-2 trial evaluated the effect of adding bosentan or placebo in functional class III or IV patients receiving intravenous epoprostenol. This study was not powered sufficiently and failed to show benefit.69 There were two studies evaluating adding inhaled iloprost to bosentan. The STEP study demonstrated safety as well as improvement in 6MWD, while a similar study conducted in Europe failed to demonstrate benefit and was terminated.70,71 The largest completed combination trial is Pulmonary Arterial Hypertension Combination Study of Epoprostenol and Sildenafil (PACES), which studied adding sildenafil as add-on therapy to intravenous epoprostenol.72 This trial enrolled 267 patients who were on stable epoprostenol therapy to receive 20mg sildenafil, titrated to 40 and 80mg TID, at four-week intervals. At the end of the 16- week study period, more than 80% of patients had reached the 80mg TID dosing level. The treated group showed a significant increase of 29m in the 6MWD, as well as hemodynamic improvements. Clinical worsening events, defined as death, transplant, hospitalization, or an increase in epoprostenol dose, were significantly better for the treated group. Several large studies are currently under way evaluating the effect of combining different classes of oral PAH regimen.
Non-PAH Pulmonary Hypertension
The term ‘non-PAH PH’ collectively refers to PH that is associated with conditions in groups two to five in the current classification (see Table 1). Left heart disease (LHD) (Group 2) is the leading cause of PH and it is well recognized that the presence of PH and subsequent RV dysfunction are independent predictors of poor prognosis.73,74 Some PAH-specific treatments have been studied in the heart failure population with negative or neutral results, and it is difficult to determine whether the right population was targeted. Defining the population who may potentially derive benefit from pulmonary vasodilator therapy with LHD is challenging. Chronic elevation of left-sided filling pressure can result in structural abnormalities of the pulmonary vascular bed, which can lead to an increase in pulmonary artery pressures, transpulmonary gradient (TPG = mPAP – PCWP), and PVR. Interventions aimed only at reducing PCWP do not normalize the TPG, but application of pulmonary vasodilators may normalize the TPG and reduce PVR. This stage is referred to as either reversible or fixed PH, depending on the presence or absence of response to acute vasodilators, respectively. The term ‘PH out of proportion’ to LHD describes the combination of elevated mPAP, TPG, and PVR in the setting of near-normal PCWP. It is yet to be determined whether this patient population may benefit from additional treatment with pulmonary vasodilators.
The FIRST study evaluating epoporstenol in heart failure was terminated early due to increased mortality.75 Inhaled iloprost has been tested in acute administration in patients with chronic heart failure and PH in peri-operative settings.76–78 Although most showed favorable acute hemodynamic effects, no long-term data are available and the evaluations need to be interpreted with caution. Bosentan has been evaluated in the heart failure population in REACH-1, which was terminated prematurely out of safety concerns, specifically elevated hepatic transaminases observed with higher doses of bosentan.79 A larger study using lower-dose bosentan (ENABLE) was conducted, studying the effects of adding ERA to neurohormonal modulators.80 The results did not demonstrate any improvement and it was a neutral study at best. There are emerging data illustrating potential benefits of sildenafil in patients with heart failure and PH. Studies have demonstrated acute hemodynamic improvements, increase in aerobic capacity, and evidence that sildenafil may modulate adrenergic response in heart failure.81–84 The first placebo-controlled trial of 12 weeks’ duration in 34 symptomatic heart failure patients with PH has been reported.85 This study demonstrated that chronic treatment with sildenafil in heart failure patients with PH resulted in hemodynamic benefits, increase in RV ejection fraction (RVEF), and improvement in exercise capacity, as well as improved quality of life. Larger clinical trials are currently in development.
Future Directions
The challenges that lie ahead in the treatment of PAH include continued efforts to increase awareness of PAH to promote early and accurate diagnosis via right heart catheterization. Since the RV is the most critical determinant of outcome, ongoing research utilizing magnetic resonance imaging (MRI) and 3D echocardiography, which can provide accurate, quantitative measures of RV function, may provide a more efficient method of detecting the disease and assessing response to treatment. Upcoming results from current clinical trials evaluating long-term morbidity and mortality outcome in using combination therapy will enhance our understanding to best utilize these complex regimens. New therapies targeting cellular proliferative pathways are actively under investigation, bringing forth a novel approach that has the potential to complement current treatments to further improve outcome. Considering the progress made so far, where improved outcome in patients with PAH is the rule and not the exception, the goal of striving for a better future appears to be a certainty.