







Cardiovascular disease is the leading cause of death in the US.1,2 Although advances in the prevention and treatment of cardiovascular disease have contributed to a decline in mortality rates, this favorable trend has slowed over the past several years.3 Recently, however, a revival of cardiovascular drug development has introduced new treatment options to the market, with several promising therapies in various stages of preclinical and clinical development. In this review, we highlight important updates in the field of cardiovascular pharmacotherapy. We focus on currently available treatments but, given the rapidly evolving landscape, include an outlook on treatments in the development pipeline. Each section focuses on a specific drug class, provides an overview of the mechanism of action and outcomes, and finishes with a practical guide to their clinical application. Key considerations are summarized in Table 1.
Proprotein Convertase Subtilisin-Kexin Type 9 Inhibitors
Overview
Proprotein convertase subtilisin-kexin type 9 (PCSK9) marks low-density lipoprotein cholesterol (LDL-C) receptors (LDL-R) on the hepatocyte membrane for intracellular degradation and thereby regulates circulating levels of the atherogenic LDL-C.4 Two fully human monoclonal antibodies against PCSK9, evolocumab and alirocumab, have been approved for use in the US. The development of a third, humanized antibody, whose amino acid sequence retained 3 % of the murine sequence, was discontinued due to detection of neutralizing antibodies in a phase 3 outcomes trial.5 A small interfering RNA (siRNA) inhibitor6 and an AT04A vaccine7 promise more durable and long-lasting anti-PCSK9 effects than monoclonal antibodies, but remain early in development.
PCSK9 monoclonal antibodies reduce circulating LDL-C by 60 % on average, in addition to their beneficial effects on other atherogenic lipoprotein species, such as lipoprotein(a), in patients without familial hypercholesterolemia. Depending on the genotype, PCSK9 inhibitors also have varying degrees of lipid-lowering efficacy in patients with familial hypercholesterolemias (FH).8 PCSK9 inhibitors reduce LDL-C by 60 % and 30 % in patients with heterozygous and homozygous FH, respectively.9–13 These effects may be blunted in FH patients with negative/negative LDL-R genotype status.10,11
Outcomes
The effect of PCSK9 inhibition on clinical outcomes was evaluated in the FOURIER trial, which randomized 27,564 patients with atherosclerotic cardiovascular disease on maximally tolerated statin therapy and an LDL-C level of at least 70 mg/dL to receive either evolocumab, administered as 140 mg every 2 weeks or 420 mg monthly at the patient’s discretion, or placebo.14 Over a median follow-up duration of 26 months, evolocumab significantly reduced the risk of the composite of cardiovascular death, myocardial infarction, stroke, hospitalization for unstable angina, or coronary revascularization by 15 % compared with placebo (9.8 % versus 11.3 %; hazard ratio [HR] 0.85; 95 % CI [0.79–0.92]; p<0.001). The composite of cardiovascular death, non-fatal myocardial infarction, or non-fatal stroke was reduced by 20 % in evolocumabtreated patients compared with placebo (5.9 % versus 7.4 %; HR 0.80; 95 % CI [0.73–0.88]; p<0.001).
These reductions in the primary and secondary composite endpoints were driven by significant reductions in both non-fatal myocardial infarction and non-fatal stroke. Cardiovascular death was not significantly different between the two treatment arms, although the short followup duration may not have allowed for detection of cardiovascular deaths averted. A second cardiovascular outcomes trial using alirocumab has completed enrollment and is in long-term follow-up (clinicaltrials.gov NCT01663402).
PCSK9 inhibitors are generally well tolerated. In FOURIER, injection site reactions were infrequent but did occur in significantly more evolocumab patients (2.1 % versus 1.6 %; p<0.001). Neutralizing antibodies were not detected in any evolocumab-treated patient in FOURIER.
The neurocognitive effects of extremely low LDL-C levels are an area of interest, given the lack of available data. In the dedicated neurocognitive substudy of FOURIER (Evaluating PCSK9 Binding Antibody Influence on Cognitive Health in High Cardiovascular Risk Subjects or EBBINGHAUS), 1,974 patients underwent prospective cognitive function testing.15 The authors reported no significant differences in neurocognitive function between evolocumab and placebo. A Mendelian randomization study of Copenhagen Heart Study did not find a significant association between LDL-C level and the risk of Alzheimer’s disease, vascular dementia, or Parkinson’s disease in patients with life-long low LDL-C levels.16 Long-term follow-up of patients treated with anti-PCSK9 monoclonal antibodies will provide further reassurance to patients and clinicians. It is important to note that Mendelian randomization studies also have suggested an increased risk of developing type 2 diabetes mellitus for patients with the metabolic syndrome – an effect similar to that observed with statins.17
Practical Considerations
Evolocumab and alirocumab are currently indicated for patients with established cardiovascular disease or FH who require additional lipid lowering beyond that achievable with maximally tolerated statin therapy.
Evolocumab can be dosed subcutaneously either every other week (140 mg) with a pen injector or once monthly (420 mg). The 420 mg evolocumab dose can be administered as three successive subcutaneous pen injections or as a single, 9-min subcutaneous infusion using a dedicated single-use auto-infuser. Alirocumab dosing should begin with 75 mg every other week subcutaneously and, if necessary to achieve further LDL-C reduction, increased to 150 mg every other week. Both evolocumab and alirocumab must be stored in the refrigerator and warmed to room temperature prior to administration. Patients should be counseled about possible injection-site reactions and flu-like symptoms. LDL-C level should be checked 4–8 weeks after starting therapy, as 1–2 % of patients may be non-responders, possibly due to an undetected PCSK9 gain-of-function gene variant or non-adherence.
The central issue related to PCSK9 monoclonal antibody use is cost. The current out-of-pocket cost for either alirocumab or evolocumab approaches US$14,000 annually and cost-effectiveness analyses from the societal perspective have not found PCSK9 inhibitors to be favorable.18,19 Many insurance plans consider PCSK9 inhibitors to be Tier 3 medications, leaving patients with a significant co-pay. Patient assistance programs are sponsored by the manufacturers of alirocumab and evolocumab and can assist both clinicians and patients in navigating the insurance prior authorization approval process. These programs may also defray out-ofpocket costs for eligible patients.
Ezetimibe, an orally available inhibitor of the gut cholesterol transport protein Niemann-Pick C1-like 1 protein, reduces LDL-C by approximately 20–30 % and modestly reduces the risk of cardiovascular outcomes without significant adverse effects. Ezetimibe may be considered for patients who choose to avoid subcutaneous administration of monoclonal antibodies or for whom the cost of a PCSK9 antibody is prohibitive. Ezetimibe went off patent in the US in December 2016 and the 6 month exclusivity period for the first generic expired in June 2017. With more generics entering the US market, it is anticipated that the cost will decrease significantly and rapidly in late 2017 or early 2018. PCSK9 inhibitors should be considered preferred over ezetimibe, provided patients are comfortable with the route of administration and can afford the difference in cost. A recent simulation analysis of a large claims database found that maximal statin therapy achieves goal lipid levels in the majority of patients and reinforces the need to maximize statin therapy prior to the addition of costly non-statin lipidlowering therapy.20
Sodium–Glucose Co-Transporter-2 Inhibitors
Overview
The sodium–glucose co-transporter-2 (SGLT-2) is a protein that is responsible for up to 90 % of the sodium and glucose reabsorption in the proximal convoluted tubule of the nephron.21 The plasma glucose threshold that inhibits SGLT-2-mediated urinary glucose reabsorption is elevated in patients with type 2 diabetes mellitus. Thus, SGLT-2 inhibition induces glucosuria, natriuresis, and osmotic diuresis, which together decrease plasma glucose concentrations and lower blood pressure. SGLT-2-mediated effects level off after an initial profound response. SGLT-2 inhibitors also produce weight loss that is sustained over several years of treatment.22,23 Notably, SGLT-2 inhibitors have modest effects on hemoglobin A1c level, with a mean reduction of 0.3-0.4 % over 52 weeks. Most important, SGLT-2 inhibition does not cause hypoglycemia, unlike other glucose-lowering agents such as insulin and sulfonylureas.
Outcomes
Two major clinical trials have demonstrated the beneficial effects of SGLT-2 inhibitors in patients with type 2 diabetes mellitus. The EMPA-REG OUTCOME trial compared empagliflozin against placebo added to optimal medical therapy in patients (n=7,028) with type 2 diabetes mellitus and established cardiovascular disease.22 In contrast, the CANVAS trial enrolled patients (n=10,142) with type 2 diabetes mellitus and either established cardiovascular disease or risk factors for cardiovascular disease and randomized them to canagliflozin or placebo added to optimal medical therapy.23
In both EMPA-REG OUTCOME and CANVAS, SGLT-2 inhibition reduced the risk of cardiovascular death, non-fatal myocardial infarction, or nonfatal stroke by 14 % (EMPA REG OUTCOME: HR 0.86; 95 % CI [0.74–0.99]; p=0.04 for superiority; CANVAS: HR 0.86; 95 % CI [0.75–0.97]; p=0.02 for superiority). In both EMPA REG and CANVAS, heart failure events were reduced by nearly 40 % and additional studies will further investigate the effects of SGLT-2 inhibition in patients with heart failure and type 2 diabetes mellitus. The reader is referred to an in-depth discussion of possible mechanisms which mediate the cardioprotective effects of SGLT-2 inhibition.21
As a result of profound glucosuria induced by SGLT-2 inhibition, patients are at increased risk of genital infections. SGLT-2 inhibitors may also cause transient worsening of renal function due to prerenal volume depletion, but SGLT-2 inhibition preserves kidney function in the long term. In patients with pre-existing renal dysfunction (estimated glomerular filtration rate 45–60 mL/min per 1.73 m2), SGLT-2 inhibitors induce a lesser degree of glucosuria as a result of impaired glucose filtration but provide similar cardiovascular benefit.22,23 Reports of 20 cases of acidosis, which were described as diabetic ketoacidosis by the reporting clinician and occurred in the setting of risk factors for diabetic ketoacidosis, prompted the United States Food and Drug Administration to issue a warning that SGLT-2 inhibitors may cause diabetic ketoacidosis.24
In the CANVAS trial, amputations of the toes, feet, or legs occurred at a rate of 0.6 events per 100 patient-years in canagliflozin patients compared with 0.3 events per 100 patient-years in placebo patients (p<0.001). Seventy-one percent of amputations were of the toe or metatarsal. The frequency of amputations was significantly greater in patients with prior amputations or peripheral vascular disease, although the effects of canagliflozin on amputations were not significantly different across these subgroups. Based on these findings, the United States Food and Drug Administration issued a boxed warning that canagliflozin increases the risk of leg and foot amputations.25 The risk of amputation did not appear to differ between empagliflozin and placebo in EMPA-REG OUTCOME.
Practical Considerations
SGLT-2 inhibitors should be considered second-line therapies to metformin for patients with established or elevated risk for cardiovascular disease and type 2 diabetes mellitus.26 Patients with low blood pressure or orthostatic hypotension, severe renal impairment (estimated glomerular filtration rate <30 mL/min per 1.73 m2) or a history of urinary tract infections or genital infections should not receive an SGLT-2 inhibitor. Canagliflozin should be avoided in patients with a history of lower extremity amputation or peripheral vascular disease until further data are available. As evidence accumulates, SGLT-2 inhibitors may become first-line or co-first-line with metformin in patients with both established cardiovascular disease and type 2 diabetes mellitus.
Dosing for both empagliflozin and canaglifozin should start at the lowest available dose (10 mg once daily and 100 mg once daily, respectively) and subsequently increased to 25 mg for empagliflozin or 300 mg for canagliflozin as needed to achieve the goal hemoglobin A1c level. The addition of an SGLT-2 inhibitor may decrease the need for glucose-lowering therapies and therefore the doses of insulin and sulfonylurea should be lowered as a precaution due to their hypoglycemic effects.
Patients should be assessed for symptoms of urinary tract or genital infections while taking empagliflozin or canagliflozin. Periodic assessment of serum creatinine is warranted, with greater frequency in patients with baseline impairment in renal function. At the time of SGLT-2 inhibitor initiation, a decrease in the loop diuretic dose should also be considered, if applicable, to decrease the possibility of volume depletion. Out-ofpocket costs for these agents can exceed several hundred dollars per month without insurance coverage.
Glucagon-like Peptide-1 Receptor Agonists
Overview
Glucagon-like peptide-1 (GLP-1) is an endogenous incretin that inhibits glucagon secretion, increases insulin secretion in response to postprandial or fasting hyperglycemia, slows gastric emptying, and increases satiety.27 GLP-1 receptor agonists decrease hemoglobin A1c levels by 0.3–1.4 % in addition to having weight-lowering and blood pressurelowering effects. A high dose of liraglutide is marketed with an indication for weight loss.28–30 GLP-1 agonists require hyperglycemia to exert insulinotropic effects and therefore cannot cause hypoglycemia.
Outcomes
Long-acting GLP-1 agonists decrease cardiovascular outcomes in patients with established cardiovascular disease or risk factors for cardiovascular disease and concomitant type 2 diabetes mellitus. In the LEADER trial, 9,340 patients received once-daily liraglutide or placebo for a median duration of 3.8 years.31 Liraglutide significantly reduced the risk of cardiovascular death, non-fatal myocardial infarction, or nonfatal stroke by 13 % (13.0 % versus 14.9 %; HR 0.87; 95 % CI [0.78–0.97]; p=0.01 for superiority). In the SUSTAIN-6 trial, once-weekly semaglutide was compared against placebo in 3,297 patients with established cardiovascular disease or risk factors for cardiovascular disease and concomitant type 2 diabetes mellitus.32 Semaglutide met pre-specific criteria for non-inferiority with respect to cardiovascular death, non-fatal myocardial infarction, or non-fatal stroke (6.6 % versus 8.9 %; HR 0.74; 95 % CI [0.58–0.95]; p<0.001 for non-inferiority). In addition, a post-hoc analysis demonstrated that the effect of semaglutide on cardiovascular outcomes achieved statistical significance compared with placebo (p=0.02 for superiority).
The ELIXA trial tested the hypothesis that lixisenatide, a short-acting GLP-1 agonist, reduces cardiovascular outcomes in patients with recent acute coronary syndrome and type 2 diabetes mellitus.33 Lixisenatide was neutral with respect to the primary outcome and each secondary outcome. The weaker effects of lixisenatide on hemoglobin A1c, weight loss, blood pressure, and heart rate as compared with liraglutide or semaglutide suggest that the results of ELIXA can be attributed to differences between short- and long-acting GLP-1 agonists (for a detailed review of GLP-1 biology and receptor pharmacology, see Meier).27
Practical Considerations
Although the starting dose in the LEADER trial was 1.8 mg once daily, clinicians should consider starting liraglutide at 0.6 mg once daily and then increasing, as tolerated by the patient, by 0.6 mg weekly to 1.8 mg daily to improve gastrointestinal tolerability. Semaglutide is not yet commercially available in the US.
In general, GLP-1 agonists are well tolerated. Up to 40 % of patients may experience gastrointestinal side effects, such as nausea, vomiting, diarrhea, cramping, and flatulence. Patients should be counseled that these symptoms dissipate over several weeks. Dose de-escalation and re-challenge may be required for patients who experience gastrointestinal side effects. Sulfonylurea doses should be decreased by 50 % prior to starting a GLP-1 agonist, if applicable. Cautious use of GLP-1 agonists is warranted in patients with systolic heart failure based on the findings of a phase 2 clinical trial.34
Potassium-Lowering Agents
Overview
Hyperkalemia is frequently encountered in the management of patients with cardiovascular disease, in particular those with cardiovascular and renal disease, and is associated with an increased risk of adverse outcomes.35 Moreover, hyperkalemia is a dose-limiting side effect of many life-saving cardiovascular therapies, such as angiotensin-converting enzyme inhibitors and aldosterone antagonists. Hyperkalemia management strategies include dietary interventions, adjustment of hyperkalemia-causing medications, treatment with sodium polystyrene sulfate (SPS) and, in urgent situations, insulin– glucose therapy or hemodialysis. SPS is an orally administered sodium– potassium exchange resin that has been associated with significant gastrointestinal discomfort and risk of bowel obstruction, as the resin swells upon contact with water. Due to these adverse effects, SPS is not tolerable as a chronic-use medication.
Patiromer is a cation-exchange polymer that is reconstituted in water to create an orally available potassium binder.35 Patiromer exchanges calcium ions for potassium ions in the distal colon without the adverse effect profile of SPS. Sodium zirconium cyclosilicate is a sodium–potassium exchanger that acts throughout the gut.35 ZS-9 has yet to be approved for use in the US due to manufacturing and production issues. Comparative data for patiromer and ZS-9 are not available.
Outcomes
Patiromer has been shown to control potassium levels within a safe range in patients with hyperkalemia who were receiving renin– angiotensin–aldosterone system inhibitors. Time to first episode of hyperkalemia was significantly longer in patiromer-treated patients compared with control. The potassium-lowering effects of patiromer appear to be related to the baseline serum potassium level, with potassium-lowering effects greatest among those with the highest baseline potassium level. In the OPAL-HK trial, mean serum potassium decreased by 1.23 ± 0.04 mmol/L among patients with baseline serum potassium levels of 5.5–6.5 mmol/L compared with a decrease of -0.65 mmol/L among patients with baseline serum potassium levels of 5.1–5.5 mmol/L.36
In patients with heart failure who were starting treatment with an aldosterone antagonist, patiromer blunted the increase in serum potassium levels, which were 0.45 mmol/L higher in the placebo arm than patiromer after 8 weeks of treatment.37 A greater proportion of patiromer-treated patients were titrated to maximal spironolactone dose than placebo (91 % versus 74 %; p=0.02).
Practical Considerations
Whether novel potassium-lowering agents have a role in routine clinical practice is unclear. The available evidence suggests that potassiumlowering agents may facilitate up-titration of renin–angiotensin–aldosterone system inhibitors. However, these data are derived from small sample sizes. In addition, side effects of potassium-lowering agents, including hypomagnesemia, may preclude tolerability in many patients.
Monoclonal Antibody Antidote for Dabigatran
Although direct oral anticoagulants (DOACs) provide an improved safety profile and greater ease of use than vitamin K antagonists, the risk of major bleeding is 1.6–3.6 % per year.38,39 To date, four reversal agents have been developed to improve the safety margin of DOACs. The use of indirect reversal agents, such as coagulation factor replacement, has been reviewed in detail elsewhere.40
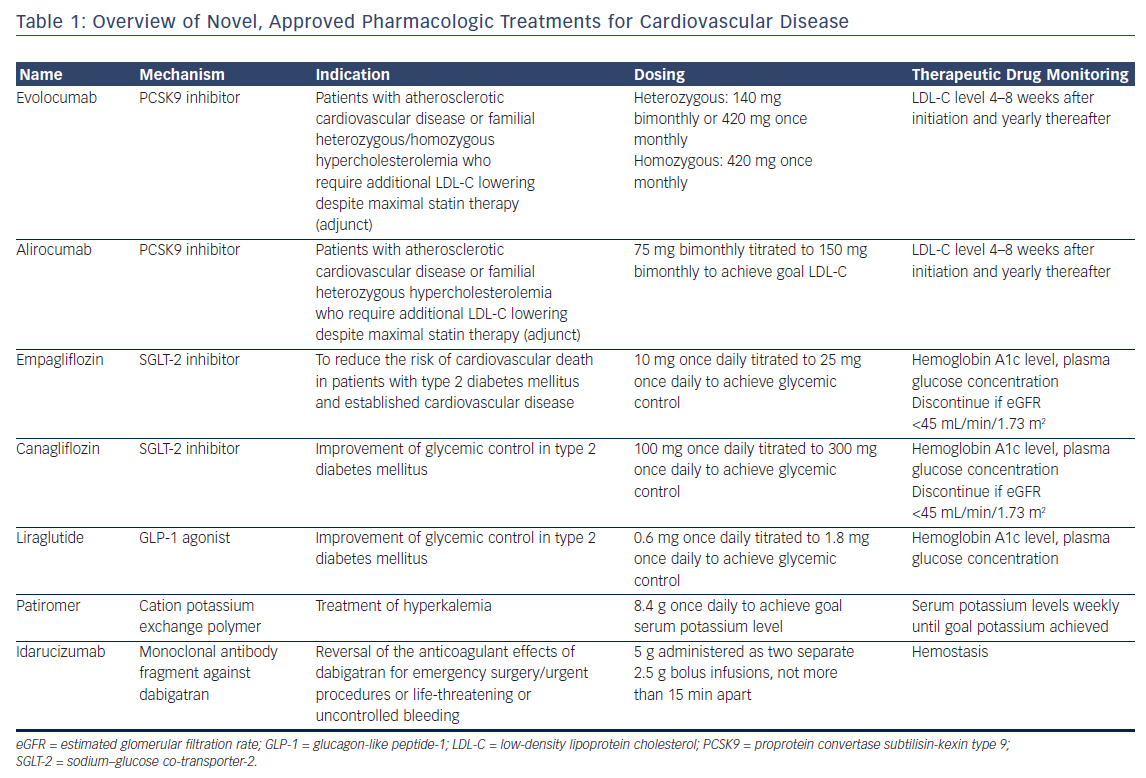
Idarucizumab is a humanized, murine monoclonal antibody Fab fragment that binds free and thrombin-bound dabigatran, an oral direct thrombin inhibitor.41 Idarucizumab’s affinity for dabigatran is 350 times greater than the affinity of dabigatran for thrombin. In the single-arm RE-VERSE AD trial, patients with uncontrolled or life-threatening bleeding (Group A, n=301) or who required an urgent procedure within 8 h of presentation (Group B, n=202) received two bolus infusions of idarucizumab 2.5 g within a 15 min period.42 Bleeding resolved within a median 2.5 h among Group A patients. Periprocedural hemostasis was graded as normal in 93.4 % of Group B patients. Among 24 patients (4.8 %) who experienced a thrombotic event within 30 days of idarucizumab, 16 were not taking anticoagulation or antiplatelet therapy at the time of the event (Figure 1). Idarucizumab was approved in late 2015 for marketing in the US.43
Practical Considerations
A significant amount of work remains to refine the usage of DOAC reversal agents as well as ensure the successful development of pipeline compounds. Reversal agents are best reserved for life-threatening bleeding events or patients who require urgent surgery that cannot be delayed until the DOAC is cleared from circulation. The decision to utilize a reversal agent should be based on the location and severity of bleeding, as well as factors that influence the drug clearance, such as renal function, time of last dose, or drug–drug interactions. Patients who have non-life-threatening bleeding, have taken the last dose 3–4 halflives prior to presentation, and have preserved renal function are unlikely to benefit from a reversal agent. In contrast, patients with intracranial hemorrhage who took a dose within a few hours of presentation or who have acute kidney injury are good candidates for a reversal agent. Idarucizumab is indicated for patients who require urgent surgery, but andexanet alfa (see In the Pipeline) was not studied in this population and is unlikely to be approved for this indication.
In the Pipeline
Interleukin-1 Blockers
Interleukin-1 (IL-1) is a cytokine regulator of the inflammatory response to myocardial insult and injury.44 IL-1 has been implicated in the pathogenesis of a wide range of cardiovascular diseases, including atherosclerotic cardiovascular disease, heart failure, and pericarditis.45,46
In the CANTOS trial, 10,061 patients with recent myocardial infarction and evidence of systemic inflammation, defined as a C-reactive protein level of at least 2 mg/L, were randomized to either canakinumab, a human monoclonal antibody directed against the beta isoform of IL-1, or placebo. Patients received either 50, 150, or 300 mg once every 3 months. The primary endpoint of non-fatal myocardial infarction, non-fatal stroke, or cardiovascular death was reduced by 15 % in the canakinumab 150 mg-treated patients compared with placebo (HR 0.85; 95 % CI [0.74–0.98]; p=0.021). In the 300 mg canakinumab group, the primary endpoint was similarly reduced (HR 0.86; 95 % CI 0.75–0.99; p=0.031), but this difference was not considered statistically significant after multipletesting adjustments. There was no significant difference in the primary endpoint between the 50 mg canakinumab group and placebo.
Other notable results from this trial include a reduction in cancer-related mortality and an increased incidence of fatal infections. IL-1 blockade does not affect T-cell function but may mask the signs of infection, such as fever, and lead to delayed diagnosis and treatment.46 Indeed, IL-1 blockade has been studied in severe sepsis with no evidence of an increased risk of mortality.47–49
The most important implication of the CANTOS trial is the opening of a new avenue of investigation for the treatment of atherosclerotic cardiovascular disease. Indeed, CANTOS is the culmination of decades of work related to the inflammatory hypothesis of atherosclerotic cardiovascular disease and may mark the beginning of a new era in cardiovascular disease research.
Cholesteryl Ester Transfer Protein Inhibitors
Cholesteryl ester transfer protein (CETP) inhibitors increase highdensity lipoprotein cholesterol and decrease LDL-C. The REVEAL trial compared anacetrapib against placebo in patients with atherosclerotic cardiovascular disease on maximally tolerated statin therapy.50 At a median follow-up duration of 4.1 years, anacetrapib-treated patients had a modest 9 % lower risk of the composite of coronary death, myocardial infarction, or coronary revascularization compared with placebo (HR 0.91; 95 % CI [0.85–0.97]; p=0.004). Considering the disappointing results of prior CETP inhibitor clinical trials,51–53 the modest benefit observed in the REVEAL trial is unlikely to lead to a change in clinical practice. In fact, it is uncertain whether the sponsor will seek marketing approval for this new therapy.54
Antithrombotic and Hemostatic Agents
Both anticoagulant and antiplatelet agents have important roles in the acute and chronic management of many cardiovascular diseases, including acute coronary syndromes and stroke prevention in atrial fibrillation. As such, the use of “triple therapy” with two platelet inhibitors and an anticoagulant has increased in recent years. Due to a lack of evidence, the optimal antithrombotic strategy in a patient with indications for both antiplatelet and anticoagulant therapy is controversial.
Several clinical trials have been designed to address this need, but additional efficacy data are required.55,56 Since many of these regimens compare dual versus triple antithrombotic therapy, reduced dose versus full dose or short versus long duration of therapy, an improved safety profile of these novel regimens is to be expected. However, these trials are not designed to thoroughly test the more uncertain question of efficacy. Also, the addition of anticoagulation to aspirin therapy in patients with stable coronary artery disease or peripheral arterial disease is an area of active research.57–59
Andexanet alfa is a recombinant protein decoy that binds direct and indirect Factor Xa inhibitors, including rivaroxaban, apixaban, edoxaban, and enoxaparin.60 Andexanet alfa lacks the catalytic domain of endogenous FXa as well as the prothrombinase complex binding domain. In the ANNEXA-4 trial, andexanet alfa, administered as a bolus followed by 2 h continuous infusion, rapidly decreased the unbound drug concentrations of rivaroxaban and apixaban.61 Thrombotic events occurred in 12 patients (18 %) within 30 days of andexanet alfa administration. Of these 12 patients, 11 were not receiving anticoagulation at the time of the event (Figure 1). Andexanet alfa has not received marketing approval in the US at the time of writing. A second recombinant Factor Xa decoy protein, which lacks the catalytic domain but retains the prothrombinase complex binding domain and therefore has prohemostatic properties, is in preclinical development.62
Ciraparantag (PER977) is a synthetic molecule that establishes noncovalent hydrogen bonds and charge–charge interactions with direct and indirect Factor Xa inhibitors, including rivaroxaban, apixaban, low molecular weight heparin, unfractionated heparin, fondaparinux, and the Factor IIa inhibitor dabigatran. Available data indicate that ciraparantag is inactive and lacks affinity for coagulation factors. Ciraparantag remains in development, but has been granted Fast Track review status by the United States Food and Drug Administration.63
Conclusions
Cardiovascular disease remains the leading cause of death worldwide. The biological mechanisms leading to cardiovascular disease are complex and span several key biological systems. Cardiovascular drug development has led to several new treatment options and avenues for investigation. At present, the introduction of several new therapies promises immediate benefits for today’s patients and advances in basic and clinical science offer hope to tomorrow’s patients.