










Long QT syndrome (LQTS) is a primary genetic and electrical disorder that causes prolongation of ventricular repolarization and increases risk for ventricular arrhythmia-mediated syncope and sudden death.1,2 LQTS is more common in women than men, even after adjustment for the longer normal QT interval in women.3 An increased and unbalanced maternal transmission of deleterious LQTS gene mutations to daughters rather than to sons contributes to the preponderance of females with LQTS.4 Importantly, post-pubertal women with LQTS are at higher risk than men for serious cardiac events, including torsades de pointes (TdP).5,6
LQTS is not uncommon, occurring in about 1 in 2,000 live births.7 Therefore, most adult cardiologists are likely to encounter women in their clinical practice with LQTS contemplating pregnancy or who are pregnant. However, there are only limited reviews guiding management of the pregnant woman with LQTS. This comprehensive review highlights important information to assist cardiologists in management of these women before, during, and after pregnancy. Substantial improvements in our understanding and knowledge regarding LQTS in areas of genetics, molecular biology and electrophysiology have made LQTS a highly manageable cardiovascular disorder. Pregnant women with LQTS benefit from multidisciplinary care from electrophysiologists, general adult cardiologists, obstetricians, maternal-fetal medicine specialists, fetal cardiologists, anesthesiologists, and genetic counselors.
Long QT Syndrome Diagnosis and Pathophysiology
After exclusion of secondary causes of QT prolongation, such as hypokalemia or drug-induced causes, a diagnosis of congenital LQTS is made on the basis of a prolonged QT interval on 12-lead ECG followed by commercially available, genetic testing for established LQTS susceptibility genes. The diagnosis of LQTS is sex specific as the QT interval is longer in post-pubertal females than males.2 A prolonged QT (QTc >460 ms for for boys and girls <15 years of age; QTc >470 ms for females and >450 ms for males >15 years of age) along with unexplained syncope is sufficient to diagnose LQTS. Congenital LQTS should also be suspected in a person with a prolonged QT in association with a family history of recurrent syncope, seizures or unexplained cardiac arrest at a young age (<30 years).8 A carrier of an LQTS pathogenic variant who is asymptomatic or lacks QT interval prolongation can also be detected with clinical genetic testing.
Pathogenic variants in up to 17 genes have been associated with LQTS.9 However, the major and most important gene subtypes are LQT1 (KCNQ1 gene), LQT2 (KCNH2 gene), and LQT3 (SCN5A gene) known as LQTS types 1, 2, and 3, respectively. LQT1 results from a loss of function mutation in KCNQ1, the gene encoding the adrenergic-sensitive, slowly activating delayed rectifier potassium channels (IKs). Cardiac events typically occur at elevated hearts rates during emotional or physical stress. Diving and swimming have been identified as potent arrhythmia triggers of lethal events in LQT1. In some cases of LQT1 where the QT is normal at rest, QT prolongation may be provoked with exercise, typically at peak heart rates. LQT2 arises from a loss of function mutation in KCNH2 (also known as the human ether-a-go-go-related gene – hERG), which encodes for rapidly activating delayed-rectifier potassium channels. The cardiac events in LQT2 are typically triggered by sudden bursts of catecholamines, such as in response to abrupt loud noises such as an alarm clock or the phone or doorbell ringing, or sudden excitement. LQT3 is due to a mutation in sodium channels (SCN5A), which disrupts fast inactivation of cardiac sodium channels during early cardiac repolarization. Most adverse cardiac events occur in LQT3 during rest or sleep. Women with LQTS types 1 and 2 are at higher risk of TdP than men with the same mutation, whereas both sexes are equally vulnerable to clinical events with LQTS type 3.5,10
Physiological Changes During Pregnancy
The dramatic physiological changes that occur during pregnancy include increased cardiac output, decreased systemic vascular resistance and enhanced chronotropy and inotropy. The effect of pregnancy on cardiac electrophysiological characteristics is less well understood. It is speculated that myocardial stretch during pregnancy due to volume changes, altered sleep cycles and circulating hormones affect cardiac ion channel function. Sex hormones modulate the clinical course of women with LQTS throughout their lifetime, and pregnancy is an important modulator of outcome in LQTS. Alteration in sex hormone levels during pregnancy and the postpartum period influences cardiac repolarization and the likelihood of clinical events in LQTS. Most studies show that the risk of LQTS-related cardiac events decreases during pregnancy compared with the time period before a woman's first conception, but increases significantly in the 9-month postpartum period, and frequency of events returns to pre-pregnancy levels after this period.11–14 The hyper-estrogenic state during pregnancy may provide protection against arrhythmias.14,15 Estrogen has been shown to downregulate expression of cardiac β-1 adrenergic receptors and reduces risk of TdP in LQTS animal models.16,17 The rapid decline in estrogen levels after parturition is needed for lactation. Estrogen withdrawal may increase adrenergic responsiveness and contribute to increased postpartum risk of cardiac events.
Prenatal Recommendations in Long QT Syndrome
Once pregnancy is confirmed in a woman with LQTS, a management plan for pregnancy and the 9-month postpartum period should be made in collaboration with the obstetrician and cardiologist with ongoing review and discussion throughout. Women should be educated and counseled on potential triggers of LQTS cardiac events including avoidance of hypokalemia and QT-prolonging drugs, which can be checked on the Credible Meds website (https://www.crediblemeds.org). Several recommendations regarding the management of pregnant patients with LQTS have been included in other guidelines.18 An ECG should be performed at each visit to evaluate the corrected QT interval. Electrolytes and vitamin D levels should be monitored as mild hypomagnesemia and vitamin D deficiency are common during pregnancy and could put the mother and fetus at avoidable risk. Encouraging increased potassium and magnesium intake is reasonable. A maternal-fetal medicine specialist with expertise in the prenatal diagnosis and care of fetal LQTS can assist parents during the prenatal period. Likewise, early referral to a pediatric cardiologist familiar with LQTS will help expedite screening of the newborn and preventive therapy in the event that the baby is diagnosed with congenital LQTS.
Management of Women with Long QT Syndrome During Pregnancy
Treatment with a β-blocker is indicated to reduce risk of cardiac events and sudden cardiac death.14,18–20 Guidelines for management of ventricular arrhythmias and prevention of sudden cardiac death strongly recommend that in women with LQTS, a β-blocker should be continued during pregnancy and the postpartum period regardless of symptoms, including while breastfeeding.18 Arrhythmic events during pregnancy are not increased among women receiving β-blocker therapy.11,12,14,19 In contrast, in a case-control study, women with LQT1 who did not receive β-blockers during pregnancy were at increased risk of cardiac arrest or syncope.13 In at least one study, increased risk for cardiac events in the high-risk postpartum period was significantly reduced by β-blockers.14
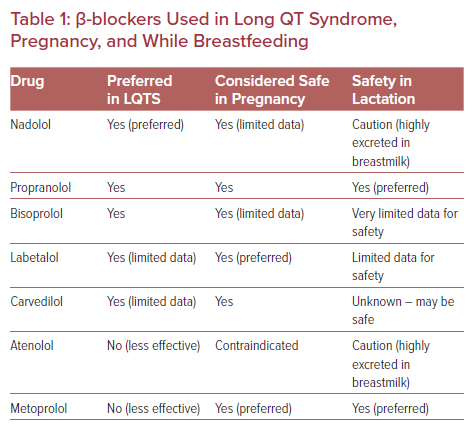
Not all β-blockers are equally effective in LQTS (Table 1).21 In general, non-selective β-blockers, such as nadolol and propranolol, are preferred over the β1-selective agents, such as metoprolol. Documented evidence, however, is largely lacking comparing specific agents in LQTS during pregnancy and lactation. Most data are primarily limited to isolated small case reports. Nadolol titrated to a recommended dose of 1–1.5 mg/kg/day is prescribed for LQTS by the majority of electrophysiologists as it is the most effective β-blocker for the syndrome, especially in high-risk individuals.22,23 The longer half-life of nadolol compared with other β-blockers also gives it an advantage for LQTS. However, data supporting the use of nadolol in pregnancy are limited. Nadolol is highly excreted in breastmilk and infants exposed to nadolol via breast milk should be monitored for side-effects, such as bradycardia, lethargy, poor feeding or weight gain. Propranolol or bisoprolol appear to have acceptable efficacy and safety profiles in LQTS and during pregnancy and lactation.24,25 Metoprolol is not advised as it is less effective in LQTS than preferred β-blockers.21 Atenolol should be avoided as it is contraindicated in pregnancy (pregnancy risk category D), due to reports of intrauterine growth restriction, and is not effective in LQTS.
β-blocker dosage should be individualized based on clinical status, tolerance, side-effects, symptoms, heart rate, blood pressure, and QT interval monitoring. The goal should be to minimize side-effects and maximize medication adherence recognizing that side-effects with higher doses may lead to non-adherence. Side-effects of β-blockers include hypotension, fatigue, and depression. However, symptoms reported during pregnancy may overlap with potential side-effects of β-blockers. Dialogue between cardiology and obstetrics during prenatal maternal assessments may be warranted to interpret symptoms and adjust medications accordingly. The pharmacokinetics of drugs may change throughout gestation. For example, β-blocker dosage may need to be increased during the later stages of pregnancy due to increasing blood volume and drug excretion. The objective should be to aim for a resting heart rate between 50 and 100 BPM. Exercise testing may be helpful in selected cases to assess the adequacy of the β-blockade with physical activity or catecholamine surges.
There are some safety concerns about the use of β-blockers during pregnancy with respect to the fetus. However, β-blockers are the most widely used cardiovascular medications and there has been a long clinical experience with these medications in pregnancy. In a large retrospective study, there was no significant increase in congenital cardiac abnormalities, after adjusting for maternal factors, among pregnant women exposed to β-blockers.26 Fetuses exposed to β-blockers may have more bradycardia and hypoglycemia and be at risk for fetal intrauterine growth restriction resulting in newborns being small for their gestational age.19,27–30 If concern for fetal growth is identified at any stage of pregnancy, prompt communication between obstetrics and cardiology is warranted. An increased risk of preterm birth was observed in some studies. Use of β-blockers during labor does not prevent uterine contractions and vaginal delivery. Overall, β-blockers are safe in pregnancy and provide protection from sudden death and syncope in mothers affected by LQTS, especially in high-risk women who have a resting QTc >500 ms, previous cardiac arrest, a high burden of ventricular arrhythmias or an ICD in place.
Antiarrhythmic drugs are generally avoided in pregnancy unless absolutely necessary, as risk to the fetus with many of these drugs remains unknown or has been inadequately studied. β-blockers are less effective in LQT3 than in other LQTS variants and sometimes sodium channel-blockers, such as flecainide or mexiletine, are used in patients with this less common LQTS variant. Flecainide has a long history of safe clinical use in pregnancy for treating maternal or fetal supraventricular tachycardia and limited data similarly suggest that mexiletine is safe in pregnancy.
Management of Long QT Syndrome During Labor and Delivery
Appropriate medical management during labor and delivery for women with LQTS includes continuous telemetry ECG monitoring for ventricular arrhythmias. Electrolyte imbalance (hypokalemia and/or hypomagnesemia) should be corrected. A 12-lead ECG should be obtained as soon as possible in women with LQTS who are in labor. A QTc >500 ms should be discussed with cardiology or cardiac electrophysiology. β-blockers should be continued on admission as they reduce the risk of serious arrhythmias. Most deliveries occur without serious arrhythmic events. However, resuscitation equipment including an external defibrillator should be easy to access during labor. Inactivating ICD therapy is not recommended in women with an ICD in place. A donut magnet should be placed over the ICD during cesarean section to avoid inappropriate shocks during electrocautery and external defibrillator pads should be placed on the thorax before preparing the patient for surgery.
QT-prolonging drugs, such as ondansetron, and certain anesthetics, such as sevoflurane, should be avoided.31–33 Oxytocin, used to induce labor and reduce bleeding, is on the list of drugs to avoid in LQTS.31 Safe vaginal delivery with use of oxytocin for the induction of labor has been reported in patients with LQTS, however cases of ventricular tachycardia developing immediately after administration of oxytocin have also been reported.34,35 In experimental models of LQT2, oxytocin and prolactin prolong QT by reducing IKs.36 Therefore, these drugs should be avoided wherever possible in high-risk LQTS women or used with due caution in the presence of telemetry ECG monitoring. If induction is performed, IV magnesium 2-4 g should be given prophylactically to reduce the risk of TdP even though magnesium can slow down labor.
Postpartum Management of Long QT Syndrome
Women with LQTS have an increased risk for cardiac events, including sudden cardiac death, in the first 9 months following delivery.11–14 In one retrospective analysis of 111 probands with LQTS, 10% experienced their first cardiac event in the postpartum period and were more likely to have multiple events.11 Probands had a 40-fold increased risk of a serious cardiac event during the postpartum period, but treatment with β- blockers was independently associated with a decrease in the risk for cardiac events. In another study, women treated with β-blockers experienced cardiac events at a rate of 0.8%, while women who were not treated with β-blockers experienced cardiac events at a rate of 3.7%.14 Therefore, it is essential for high-risk women with LQTS to continue taking β-blockers throughout the postpartum period as first-line, protective therapy for which the benefits outweigh risks of treatment.
Mothers with LQTS should be seen by a cardiologist within the first few weeks postpartum and every month for the first 9 months to ensure adherence to β-blocker therapy and to review heart rates, QTc, and symptoms. Some women who required an increase in β-blocker dose during pregnancy may need a decrease in dose later (after 6–9 months postpartum). Adequacy of β-blockade can be assessed with exercise testing or event monitoring aiming to reduce peak exercise heart rates. β-blockers are excreted in breast milk and there is potential for β-blockade in nursing infants. Domperidone, used to stimulate lactation, is a QT-prolonging drug and is contraindicated in LQTS patients.
Case Report One
A 28-year-old woman with genotyped LQT1 presented for routine cardiac evaluation. She was asymptomatic and was taking β-blocker therapy (nadolol 60 mg daily). She had a family history of LQTS and premature sudden death. Her QTC was 450 ms and in the past her QTc prolonged to more than 550 ms during stress testing at peak exercise. She was contemplating pregnancy, but in light of her cardiac history, she had concerns about the risks. She wanted to know if any precautions needed be taken to reduce the risk of adverse events and how likely it would be that her child would inherit LQTS and be at risk of cardiac events.
Preconception Counseling
Pregnancy is a major concern for women with LQTS. When a woman with LQTS is contemplating pregnancy, she and her family should be counseled and encouraged that it is likely that she will have an uneventful pregnancy and successful labor and delivery. Pregnancy will often provoke anxiety in women with LQTS and they will need support before, during and after pregnancy. Women should be reassured that LQTS is not a contraindication to pregnancy and the overwhelming majority of women with LQTS do not experience cardiac events while pregnant.
A woman with LQTS should be under the management of a cardiac electrophysiologist or cardiologist experienced in LQTS management. The importance of continuing β-blocker therapy throughout pregnancy and the postpartum period should be stressed. Assessment should be made of the risk status and effectiveness of current management. Patients with LQT1 generally have a low risk for cardiac events throughout their lifetime including during and after pregnancy and in most cases will not require an ICD for primary prevention if appropriately managed with β-blockers. If an ICD is indicated in LQTS due to high-risk status, implantation should be performed prior to pregnancy or preferably after the first trimester.18 An ICD is not a contraindication to pregnancy and there are reports of women receiving ICD shocks during pregnancy without adverse fetal outcomes.37
Prenatal genetic counseling also may be considered to discuss and explain the risk of inheritance of a pathogenic gene variant. LQTS is inherited in an autosomal dominant manner so there is a 50% chance that each child inherits the genetic variation. In rare situations where both parents have LQTS, the risk to each child is 75%. The genetic mutation does not skip generations. Families with LQTS variants should be referred to a maternal fetal specialist or pediatric cardiologist familiar with LQTS for further discussions regarding fetal LQTS diagnosis and management of LQTS in children.
Case Report Two
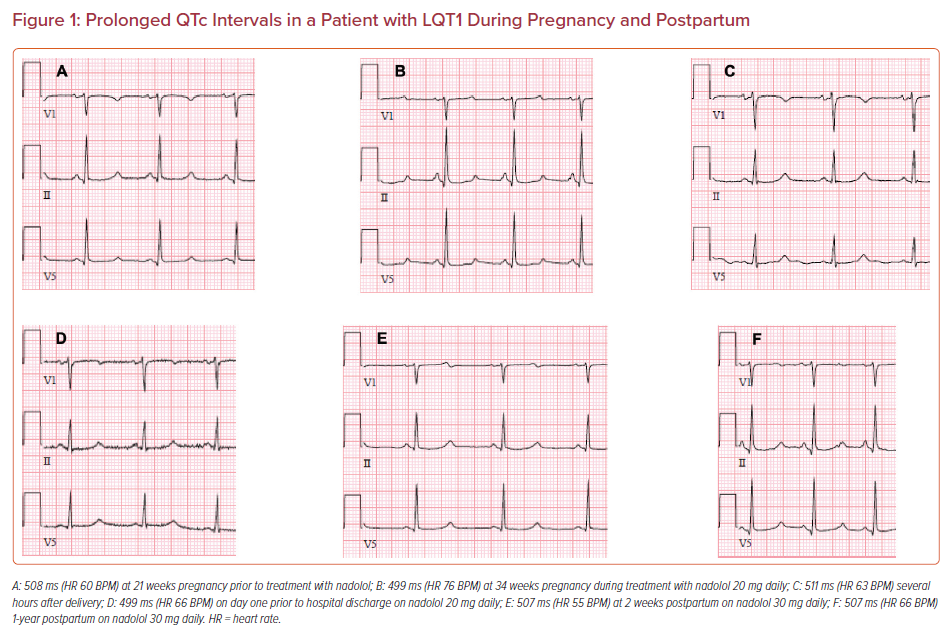
A 39-year-old woman with genotyped LQT1 was referred by her obstetrician at 21 weeks of pregnancy for cardiology evaluation. She was not taking any medications other than multivitamins and had never been prescribed β-blocker therapy. She was asymptomatic but had a family history of LQTS and sudden death in an uncle at age 40 and a sister with LQTS with an ICD implanted for aborted cardiac arrest. Her ECG showed a QT >500 ms (Figure 1). She was prescribed a β-blocker (nadolol at progressively increasing doses) with careful QT monitoring on repeated ECGs. Early referral was made to a pediatric electrophysiologist and genetic counselor to assist with screening the newborn for LQTS. She delivered a preterm baby boy with a normal birthweight just before 37 weeks. The newborn’s ECG showed a prolonged QT, which was confirmed by pediatric electrophysiology. Genetic testing identified a LQT1 mutation with the same pathogenic variant as the mother. The baby was started on oral propranolol and was later switched to nadolol. The mother had regular postpartum follow-up without any cardiac events. However, she returned after having a first trimester miscarriage the following year and also reported having had a miscarriage 10 years earlier.
Management of the Neonate in a Family with a History of Long QT Syndrome
Screening of the neonate is essential when a parent has LQTS. In an affected newborn, the risk of events including sudden infant death may be as high as 4% during the first year of life.38 Preparations should be made for potential fetal distress or arrhythmias in labor and delivery. Fetal bradycardia may be the first indication and an important predictor of LQTS.39–42 LQTS can be diagnosed in the fetus with percutaneous umbilical blood sampling or non-invasively with fetal magnetocardiography.43 Ventricular tachycardia, TdP, bradycardia, and/or intermittent or sustained second degree atrioventricular block are hallmark arrhythmias in babies with LQTS, and 10% of cases of sudden infant death syndrome as well as childhood deaths may be attributed to LQTS, which highlights the need for comprehensive screening.44–46 Neonatal genetic screening can be performed on the infant’s blood sample or on cord blood obtained at birth for the specific parental pathogenic mutation. For all newborns with a parent with LQTS, a 12-lead ECG should be obtained on day one or before hospital discharge and evaluated by a pediatric cardiologist or electrophysiologist.47 Given the known variability of QT interval during the first week of life, screening ECGs should be repeated after 3 or 4 weeks. Genetic testing evaluating for the known pathogenic variant in a family with LQTS should be performed on the baby regardless of ECG findings.
Miscarriage Risk in Women with Long QT Syndrome
Pregnant women with LQTS are at a higher risk of fetal death than the general population.48 A recent international study reported that when one parent had LQTS, miscarriages (fetal death at ≤20 weeks gestation) were twice as common (16% versus 8%) and stillbirths (fetal death at >20 weeks gestation) were 8 times more common (4.0% versus 0.5%) than the general population. Fetal death was significantly more frequent when the mother rather than the father had LQTS (24.4% versus 3.4%). In addition, mothers with LQTS delivered earlier and infants weighed less than infants of fathers with LQTS. Stillbirths were documented in fetuses without an LQTS mutation, which raises the possibility that an LQTS genetic mutation and ion channel variations in mothers may affect uterine or placental structures resulting in fetal death or growth restriction.
Case Report Three
A 37-year-old woman with a family history of LQTS was referred by obstetrics for cardiology evaluation at 32 weeks of pregnancy. Her mother and two sisters had genotype-positive LQT2. There was a family history of aborted cardiac arrest in a cousin. The woman had a previous miscarriage at 8 weeks gestation. She was asymptomatic without recent syncope. QTc at the initial visit was 486 ms and subsequent genetic testing confirmed the diagnosis of LQT2 (KCNH2 mutation). She was started on β-blocker therapy (nadolol). She presented at 40 weeks and 4 days for induction of labor and was started on oxytocin. Cardiac electrophysiology consultation was obtained and recommendations were to initiate telemetry monitoring, discontinue oxytocin, avoid ondansetron as an anti-emetic and prophylactically treat with IV magnesium. She had a successful vaginal delivery of a healthy baby girl. The mother's QTc was 487 ms on postpartum ECG at hospital discharge, while continuing to receive nadolol.
Management of High-risk Long QT Syndrome Patients
The increased risk of cardiac events in LQTS in the 9-month postpartum period is highest in subjects with LQTS with a type 2 mutation.11,14 In one study, postpartum cardiac events, including life-threatening episodes, occurred in 16% of patients with LQT2 compared with <1% with LQT1. The early postpartum period is associated with changes in hormonal balance, sleep deprivation, fatigue, stress, sudden auditory stimuli, such as a crying baby, that may be responsible for triggering and clustering of these events postpartum in LQT2. Close cardiac follow-up of women with LQT2 mutation during the postpartum period is recommended with serial ECGs every few weeks after delivery in consultation with a cardiologist experienced in LQTS management. Treatment with β-blockers at adequate doses is of paramount importance in LQT2 patients postpartum. An ICD should be considered in high-risk LQTS women who have had aborted cardiac arrest, syncope on β-blocker therapy or have a markedly prolonged QTc >500 ms with LQT2 or LQT3.18 In one study, two women with LQT2 required an ICD 4–8 weeks after delivery due to episodes of polymorphic ventricular tachycardia despite treatment with a β-blocker (metoprolol).49 Since the high-risk period might last only nine months in these patients with LQT2, another option to consider is a wearable defibrillator, but there is very limited experience with this approach during pregnancy and postpartum.
Conclusion
Women with congenital LQTS require a team approach involving cardiology and obstetrics before, during and after pregnancy to optimize care and reduce the risk of potentially life-threatening events in the mother, fetus, and baby. Cardiologists caring for women with LQTS need to be aware that the risk of cardiac events increases during the 9-month postpartum period. Treatment with a β-blocker, at an optimal dose, which is preferred in LQTS and considered safe in pregnancy along with close monitoring, is recommended during pregnancy and the high-risk postpartum period.