










Heart failure with preserved ejection fraction (HFpEF) and hypertrophic cardiomyopathy (HCM) are two distinct heart conditions that share similar clinical characteristics and diagnostic challenges. The two conditions often coexist and are commonly encountered in clinical practice.
HFpEF is defined as signs and symptoms of heart failure caused by structural/functional cardiac abnormalities with a left ventricular ejection fraction (LVEF) >50% and elevated natriuretic peptides, or objective evidence of cardiopulmonary/systemic congestion.1 The principal pathophysiological features of HFpEF are often rooted in diastolic dysfunction, a process in which there is impaired myocardial filling of the left ventricle (LV) resulting from increased ventricular stiffness, which, in turn, restricts passive LV filling, or delayed relaxation which restricts active filling.2 While diastolic dysfunction is a component of HFpEF, HFpEF is a broader clinical syndrome that is inextricable from the classic signs and symptoms of heart failure, which include dyspnea on exertion, orthopnea, and paroxysmal nocturnal dyspnea.
HCM is defined as a disorder of cardiac myocytes characterized by cardiac hypertrophy and myocardial fiber disarray disproportionate to loading stimuli (e.g. hypertension or aortic stenosis).3 This severe hypertrophy is often associated with a small LV cavity and a decrease in LV relaxation and compliance, which contribute to underlying diastolic dysfunction.4 Left ventricular hypertrophy can be also seen in uncontrolled hypertension, aortic stenosis, athlete’s heart, and infiltrative diseases including amyloidosis, sarcoidosis, hemochromatosis, and glycogen/lysosomal storage diseases, which can be challenging phenotypic mimickers of HCM.
In the US, the reported prevalence of unexplained asymptomatic hypertrophy in young adults ranges from 1 in 200 to 1 in 500.5 In the general population, the prevalence of HCM is estimated to range from 1 in 625 to 1 in 344 (0.16% to 0.29%).5 In the US, 100,000 people are diagnosed with HCM while a further 750,000 are estimated to have the condition.5 Globally, it has been estimated that HCM affects more than 20 million individuals.5
There is an unmet need to develop advances in clinical, imaging and genetic risk stratification methods to improve the recognition, understanding, and clinical outcomes of HCM. As such, it is crucial to distinguish between hypertrophic obstructive cardiomyopathy and potential mimickers, such as HFpEF or left ventricular hypertrophy (LVH), resulting from other conditions to ensure accurate diagnosis and appropriate treatment.
Clinical Manifestations
Individuals with HCM are often asymptomatic and diagnosed incidentally. Symptomatic patients commonly present with dyspnea, fatigue, chest discomfort, palpitations, presyncope and syncope, or sudden cardiac death (SCD). The mechanisms underlying dyspnea include elevated left ventricular end diastolic pressure from diastolic dysfunction, mitral regurgitation, left ventricular outflow tract obstruction (LVOTO), and ischemia.
Chest discomfort results from an oxygen demand–supply mismatch, intramural compression of small arteries from myocardial hypertrophy, vascular remodeling, and abnormal coronary flow reserve.6,7 Unexplained syncope is an established risk factor for SCD. The potential causes can be broadly divided into arrhythmias or a primary hemodynamic mechanism. The former include atrial or ventricular tachyarrhythmias or bradyarrhythmias, heart block, and sinus node dysfunction. The latter includes LVOTO, abnormal blood pressure response during exercise, and hypotension from decreased preload in the setting of diastolic dysfunction.8,9 There is considerable day-to-day variation in the severity of symptoms, with worsening of symptoms in hot or humid weather or after large meals or alcohol consumption.
On physical examination, the classic finding of LVOTO is a mid-peaking crescendo-decrescendo systolic murmur best heard at the left sternal border in patients with obstructive HCM. A separate, distinct, mid-systolic, high-pitched, blowing systolic murmur caused by mitral regurgitation, resulting from distortion of the mitral valve apparatus related to systolic anterior motion of the mitral valve, may be best heard at the apex.10 The intensity of both murmurs varies with maneuvers that alter stroke volume or LVOTO, such as standing from a squatting position or the strain phase of the Valsalva maneuver, and post-premature ventricular contraction.
In the absence of LVOTO, signs of LV hypertrophy may be identified such as brisk carotid arterial pulsation with a spike-and-dome pattern, sustained apical impulse and the fourth heart sound. Non-obstructive HCM may present with signs of volume overload due to a small restrictive LV, with elevated jugular venous pressure or lower extremity edema.
Diagnosis
Echocardiography
Transthoracic echocardiography (TTE) is the initial imaging modality of choice for the diagnosis of HCM (Table 1).3
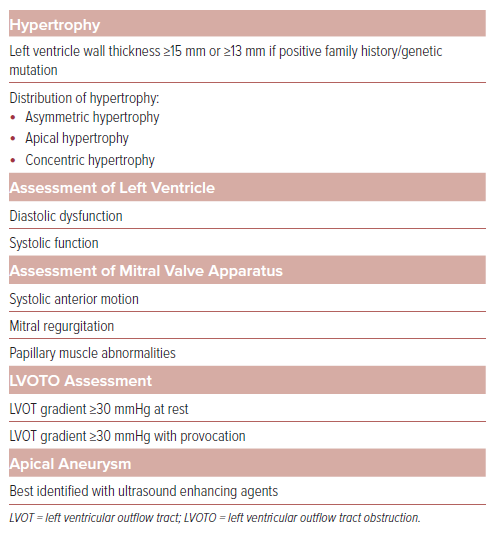
An LV wall thickness of ≥15 mm in the absence of other etiologies is diagnostic for HCM; however, alternative etiologies for LV hypertrophy must be investigated. An end-diastolic wall thickness of ≥13 mm can be diagnostic if there is a family history of HCM or a known disease-causing genetic mutation. The most common pattern of hypertrophy is asymmetric, characterized by a thickening of the proximal or basal septum of the heart.11 Other patterns of hypertrophy that may be observed include sigmoid septum, reversed septal curvature, mid-wall, apical, concentric, and right ventricular hypertrophy, and, in some rare instances, isolated hypertrophy of papillary muscles.12,13
TTE is essential for the initial assessment of the left ventricular structure and function, involving comprehensive assessments of systolic and diastolic function. LVEF should be assessed with or without ultrasound-enhancing agents at the time of diagnosis and when there is a significant change in clinical condition.
Global longitudinal strain, a novel imaging technique using speckle-tracking echocardiography, offers a complementary approach of assessing systolic function by directly measuring the myocardial deformation.14 Recent evidence suggests that strain rates are lower in HCM and that lower (less negative) global longitudinal strain values are associated with increased cardiovascular risk.15
Indices used to identify diastolic dysfunction include mitral inflow velocities recorded at the annulus and leaflet tip levels, early diastolic velocity by tissue Doppler measured at the septal and lateral sides of the mitral annulus, peak tricuspid regurgitation velocity obtained by continuous wave Doppler, left atrial volume, and pulmonary vein velocities. Left atrial volume and E/e’ ratio should be interpreted with caution in the presence of moderate or more severe mitral regurgitation (MR), as they may be affected regardless of left atrial pressure. In such cases, peak tricuspid regurgitation velocity and pulmonary vein atrial velocities are more reliable indicators of LV filling pressures. Left atrial strain is an alternative parameter that can accurately categorize LV diastolic function and predict heart failure events in HCM.16
On echocardiography, the findings typically observed with LVOTO are aortic valve mid-systolic partial closure and systolic anterior motion (SAM) of the MV with septal contact and turbulent color Doppler flow in the left ventricular outflow tract (LVOT). The severity of LVOTO can be reliably assessed using continuous and pulsed wave Doppler, taking care to avoid contamination from the intrinsic MR jet.17 LVOTO is evident by a late-peaking or dagger-shaped Doppler profile and is considered significant with a peak gradient of >50 mmHg at rest or with provocation. Provocation maneuvers should be performed for each patient, especially if normal resting gradients are observed. A provoked gradient of ≥30 mmHg is associated with an increased risk and progression to severe heart failure symptoms (New York Heart Association [NYHA] class III or IV).18
According to the 2020 American College of Cardiology/American Heart Association (AHA/ACC) guidelines for patients with HCM, exercise stress echocardiography is the preferred method for provoking a LVOT gradient due to its ability to induce the greatest physiologic response.19,20 Maneuvers such as Valsalva and squat-to-stand are useful in determining gradients in patients for whom accurate noninvasive estimation with exercise is not possible or is limited by inconsistencies in patient instruction and effort.21 Postprandial exercise may also be useful, especially in patients who experience symptoms after meals.21 Pharmacological provocation with amyl nitrite, isoproterenol or dobutamine may also be used, although amyl nitrite is not widely available.22
The main structural abnormalities contributing to LVOTO are septal hypertrophy with narrowing of the LVOT, SAM of the MV, and abnormalities of the mitral valve and subvalvular apparatus.23,24
SAM of the MV is characterized by abnormal movement of the MV leaflets into the LVOT, leading to or accentuating the LVOTO. The primary mechanism involves drag forces on the anterior mitral valve leaflet pulling it toward the LVOT. Additionally, Venturi forces created as flow enters the narrowed LVOT may contribute to its obstruction.25 The structural abnormalities of the mitral valve apparatus that predispose a patient to the development of SAM include elongated mitral valve leaflets, papillary muscle hypertrophy and hypermobility, apical/anterior displacement of papillary muscle, or insertion of an anomalous papillary muscle into the anterior mitral leaflet, and fusion with the LV free wall or septum leading to chordal-leaflet laxity.26
SAM is categorized as mild when it occurs briefly without septal contact, moderate when SAM and septal contact lasts for less than one-third of the systolic period, and severe when septal contact persists for more than one-third of the systolic period. Mid-ventricular obstruction is an uncommon morphologic variant of asymmetric HCM related to mid-ventricular hypertrophy and/or anomalous papillary muscle insertion. On echocardiography, it is identified by the hourglass-shaped appearance of the LV chamber and a mid-cavity gradient of >30 mmHg unrelated to SAM. Mid-ventricular obstruction is associated with LV aneurysms in around 3% of patients with HCM, and color Doppler reveals flow in the sequestered area and a paradoxical apex-to-base diastolic gradient.27
MR associated with HCM may be secondary to SAM or primary due to intrinsic pathology of the MV, which includes MV prolapse, chordal rupture, traumatic fibrosis of anterior MV from repeated contact with septum, significant mitral annual calcification and anomalous insertion of papillary muscle to the anterior mitral valve leaflet.28 The direction of the MR jet can be helpful in identifying the underlying cause. In SAM-related MR, an elongated anterior leaflet creates an inter-leaflet gap during systole, resulting in a posteriorly directed jet. On the other hand, a central or anteriorly directed jet should raise suspicion for an intrinsic valve abnormality, although in some cases SAM may be associated with a central or anterior jet in the absence of intrinsic valve pathology. Transesophageal echocardiogram (TEE) or cardiac MRI may be needed for better evaluation of the mechanism of MR before surgical correction.
HCM is associated with an increased risk of SCD and echocardiography plays a key role in risk stratification based on the recent ACC/AHA HCM guidelines. High-risk features, such as increased LV maximal wall thickness, LV aneurysm, and reduced LVEF, can be evaluated by echocardiography. If a patient exhibits at least one of these risk factors, consideration of an ICD is warranted.20
Echocardiography plays a key role in guiding treatment of HCM. The 2020 ACC/AHA HCM guidelines recommend considering intraoperative TEE for surgical myectomy and adjunctive repairs to assess three key structural changes: abnormalities in MV including SAM; extent of septal hypertrophy; and LVOTO.29–31 TTE and TEE play multiple roles in guiding successful alcohol septal ablation (ASA) by locating the appropriate septal perforator, monitoring LVOT gradient reduction, and enhancing procedural success with ultrasound agents.32 Following septal reduction therapy (SRT), echocardiography is instrumental in assessing treatment outcomes by measuring septal thinning and LVOT gradients.33
Cardiac MRI
In patients with HCM with diastolic heart failure, cardiac MRI is a helpful tool for assessing the structural changes associated with HFpEF and differentiating HCM from phenotypic mimics such as athlete’s heart, hypertensive heart disease and infiltrative cardiomyopathies (Table 2). Diastolic function parameters that can be assessed include LV mass and hypertrophy, left atrial size and function, mitral inflow and pulmonary venous velocity profiles, and myocardial deformation imaging with strain.34
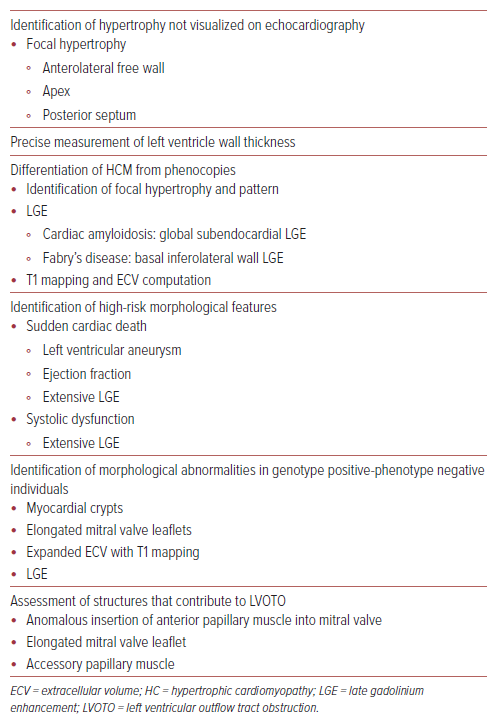
Novel cardiac MRI sequences, such as T1 mapping and extracellular volume computation, enable the direct assessment of replacement and interstitial fibrosis through measurement of longitudinal tissue proton relaxation times after an electromagnetic pulse; this is hypothesized to contribute significantly to impaired cardiac relaxation and stiffness in HFpEF. This facilitates tissue characterization and early detection of myocardial disease, including cardiomyopathies in which native T1 values and myocardial extracellular volume are increased.34 Findings on cardiac MRI that favor a diagnosis of HCM over athlete’s heart include a focal pattern of hypertrophy and unchanged maximum LV wall thickness after forced deconditioning and late gadolinium enhancement (LGE).35
Cardiac MRI has the advantage of having a high spatial resolution, which allows better visualization of focal areas of LVH, especially when localized to the anterolateral wall, overcoming limitations caused by poor acoustic windows from pulmonary or thoracic parenchyma. In hypertensive cardiomyopathy, a concentric pattern of hypertrophy is observed on cardiac MRI whereas, in HCM, it is commonly asymmetric. The presence of LVOTO is rare in hypertensive cardiomyopathy and a regression of LV wall thickness on serial measurements with intensive antihypertensive treatment may be observed.36
In a recent study, extracellular volume was found to be effective in differentiating HFpEF from hypertensive cardiomyopathy, and was superior to echocardiographic global longitudinal strain.37 The pattern and distribution of LGE contrast on cardiac MRI can help differentiate HCM from infiltrative cardiomyopathy. Increased LV wall thickness in the lateral wall and septum accompanied by subendocardial LGE on cardiac MRI is characteristic of amyloidosis.36 Conversely, LGE confined to the basal inferolateral wall may raise suspicion of Fabry’s disease, an X-linked lysosomal storage condition that results in excessive lipid deposition in tissues.38 The pattern of LGE distribution in HCM is diverse and cannot be used alone for differentiation; one must take into account the entire clinical picture to accurately distinguish HCM from other cardiomyopathies.
Cardiac MRI accurately identifies high-risk morphological features predictive of SCD predisposition and potentially life-threatening ventricular arrhythmias, including apical aneurysm, extensive LGE distribution involving multiple LV segments, massive LVH, and systolic dysfunction (EF <50%).39 Recognition of high-risk features associated with SCD can help guide decision-making for prophylactic ICD implantation.
Cardiac MRI also helps in guiding further management decisions by delineating the LVOT anatomy, which allows clinicians to determine the most suitable approach when considering SRT.40 When planning surgical intervention, cardiac MRI aids in quantifying the extent of tissue necrosis or viability and scarring in patients with long-standing, untreated HCM. It assists in pre-procedural planning by defining the cardiac anatomy, MV structure and sub-mitral structures. In HCM patients with elongated mitral leaflets, preoperative cardiac MRI enables decision-making regarding leaflet repair.40 Cardiac MRI can also be used to predict and monitor LV mass regression after alcohol septal ablation.40
CT Imaging
While cardiac CT can provide clear visualization of the LV structure in cases of HCM, it is not recommended as the primary diagnostic tool as per the 2020 ACC/AHA HCM guidelines.20 This is mainly because cardiac CT has some drawbacks, including exposure to ionizing radiation, the need for iodinated contrast, and inferior temporal resolution when compared with echocardiography. It is often reserved for specific situations when additional information beyond echocardiography and cardiac MRI is required, such as in the assessment of coronary arteries, including stenosis or anomalous origin of coronaries.27
Angiography or Invasive Assessment
Invasive hemodynamic studies aid in clarifying LVOT obstruction in patients with dynamic murmurs and/or symptoms suggestive of HCM, but who have an LVOT gradient of <50 mmHg at rest and with provocation on echocardiography.
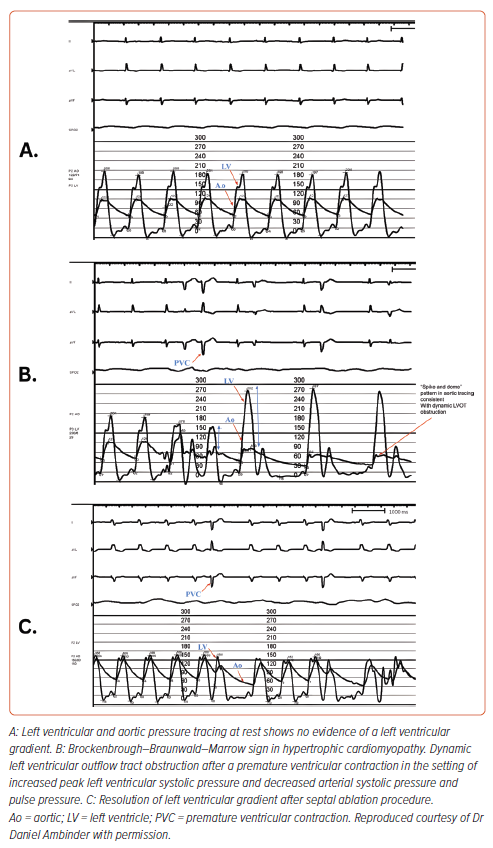
Provocative maneuvers during invasive hemodynamic studies include Valsalva, isoproterenol infusion, and inducing a premature ventricular contraction to assess for the presence of the Brockenbrough-Braunwald-Morrow sign. The latter is characterized by post-extrasystolic augmentation in the LVOT gradient and decrease in aortic pulse pressure, resulting in a spike-and-dome pattern on aortic tracings (Figure 1).41,42
Invasive assessment may also be appropriate for coexistent valvular aortic stenosis and suspected coronary artery disease as well as for those undergoing myectomy or alcohol septal ablation.
Treatment and Management
In asymptomatic patients, the management of HCM should focus on lifestyle modification, which includes maintaining adequate hydration in patients without evidence of fluid overload, avoidance of caffeine and alcohol, a balanced diet, and physical activity.
Patients should be counseled to avoid large meals and reduce levels of post-prandial activity to help control the worsening of symptoms from splanchnic blood flow sequestration followed by a decrease in circulating plasma volumes and resultant increase in LVOTO.43 Dehydration should be avoided because a decreased preload causes increased contractility and possible worsening of dynamic obstruction.
Caution should be exercised during consumption of alcohol because it decreases arterial blood pressure, and increases SAM and intraventricular obstruction.44
Pregnancy requires special considerations and care driven by a multispecialty cardio-obstetric heart team. Maternal mortality is higher in women with HCM than in those without the condition, but absolute maternal mortality is low and confined to those with high-risk features, including pre-existing heart failure symptoms, pulmonary hypertension, and severe LVOT obstruction.45 Vaginal delivery is the preferred delivery option. Selected β-blockers can be considered for symptoms related to obstruction or arrhythmias with monitoring of fetal growth. However, the cardiac myosin inhibitor (CMI) mavacamten is contraindicated, due to potential teratogenic effects.46
Pharmacological Management
The primary goal of pharmacological treatment in HCM is to alleviate symptoms due to LVOTO. Pharmacological therapy for HCM includes β-blockers, non-dihydropyridine calcium channel blockers, disopyramide, angiotensin-converting enzyme inhibitors (ACEis)/angiotensin receptor blockers (ARBs), and a newer class of drugs, CMIs (Supplementary Table 1). β-blockers are the mainstay of treatment and reduce LVOT obstruction, angina, dyspnea, and arrhythmia risk.46
Non-dihydropyridine calcium channel blockers, such as verapamil and diltiazem, can also be used because of their negative inotropic and chronotropic properties, which result in improved diastolic filling, reduced outflow obstruction, and improved subendocardial ischemia. However, current guidelines caution against their use in severe obstruction because of their potential to cause vasodilation, and in heart failure because of their negative inotropic effects.20
There is limited data on the efficacy of ACEis and ARBs in HCM. The safety and efficacy of the ARB, valsartan in attenuating disease progression in early HCM was assessed in the VANISH trial, a multi-center, placebo-controlled, Phase II randomized control trial. A total of 178 participants 8–45 years of age with non-obstructive HCM with pathogenic or likely pathogenic sarcomeric variants, NYHA class I or II symptoms, normal EF, no secondary prevention ICDs, and no history of appropriate ICD shocks or prior SRT were randomized 1:1 to valsartan or placebo for 2 years. A significant attenuation in the composite end point of LV wall thickness, LV mass, LV volume, left atrial size, diastolic parameters, and biomarkers was observed in the valsartan group (between group difference 0.231, 95% CI [0.098–0.364]; p=0.001).47 There has been no proven benefit of losartan to date; however, it is safe and may be used for other indications in patients with HCM.48
The 2024 AHA/ACC HCM guidelines recommend the use of CMI, disopyramide and septal reduction therapy (SRT) in patients with persistent symptoms despite first-line therapy.46 Disopyramide has been shown to reduce outflow gradients and improve symptoms in patients with outflow obstruction.49 However, it should be used in combination with a atrioventricular nodal blocking agent, such as a β-blocker or calcium channel blocker, because this drug can increase the risk of AF with rapid ventricular rate by enhancing conduction through the atrioventricular node
CMIs are a novel class of drugs that act by blocking myosin ATPase, which reduces the availability of myosin heads for engagement in cross-bridge formation with actin filaments. This leads to a reduction in myocardial hypercontractility, relieving LVOT obstruction, decreasing wall stress and improving the rate of myocardial relaxation.50, 51
Mavacamten is the first-in-class CMI approved for treatment of obstructive HCM. This was initially presented in the PIONEER-HCM study, an open-label, non-randomized trial, where mavacamtem demonstrated effectiveness in reducing the LVOT gradient and improving peak oxygen consumption (pVO2), the latter indicated by increased exercise capacity.51
This was further studied in EXPLORER-HCM, a Phase III, multi-center, double-blind trial that randomized 251 patients with HCM with an LVOT gradient of >50 mmHg and NYHA class II–III symptoms to mavacamtem starting at 5 mg or placebo for 30 weeks.52 Mavacamten doses were adjusted in a two-step blinded titration approach to achieve target reduction in an LVOT gradient of <30 mmHg and mavacamten plasma concentration of 350–750 ng/ml. The primary endpoint was a composite to assess clinical status at week 30, which was defined as: a 1.5 ml/kg/minute or greater increase in pVO2 and at least one NYHA class reduction; or a 3.0 ml/kg/minute or greater improvement in pVO2 and no worsening of NYHA class. At the end of the study, 37% of patients in the mavacamten group met the primary endpoint (difference 19.4%; 95% CI [8.7–30.1]; p=0.0005) versus 17% of patients in the placebo group. Furthermore, secondary endpoints including post-exercise LVOT gradient, pVO2, and Kansas City Cardiomyopathy Questionnaire symptom score were significantly improved in mavacamten compared to placebo. Seven patients in the mavacamten group and two in the placebo group had a transient decrease in LVEF to <50%, with three on mavacamten (two in the placebo arm) requiring protocol-driven discontinuation of the drug. No major cardiovascular events occurred.
These results were echoed in the follow-up study, VALOR-HCM, a multicenter, Phase III, double-blind, placebo-controlled study that focused on patients with symptomatic obstructive HCM, despite maximally tolerated medical therapy, who were eligible to undergo SRT.53,54 Results indicated that a majority of patients on mavacamten no longer met guideline criteria for SRT after 16 weeks (difference 58.9%; 95% CI [44.0%–73.9%]; p<0.001) or 32 weeks; a similar effect was observed in patients who crossed over from placebo after 16 weeks. The study also showed markedly decreased post-exercise LVOT gradient and improvements in NYHA functional class of one or more, as well as better patient-reported outcomes (9.4 points).
Aficamten is a next-in-class CMI being studied for the treatment of obstructive HCM. Initial results from REDWOOD-HCM, a Phase II randomized, placebo-controlled, double-blind study, indicated similar outcomes to mavacamten, with improvement in LVOT obstruction and NYHA class with no adverse events requiring termination of therapy or treatment interruptions.55
These findings are supported by SEQUOIA-HCM, a phase III randomized, placebo-control trial in adults with symptomatic HCM, which showed that treatment with aficamten led to significantly greater improvement in peak oxygen uptake, LVOT gradients and fewer days eligible for SRT as compared to placebo. The ongoing MAPLE-HCM trial (NCT05767346) will be the first to provide a head-to-head clinical comparison of CMI with β-blockers in patients with obstructive HCM.
While CMIs have been proven to be a benefit in the treatment of obstructive HCM, in clinical practice patients receiving them require periodic echocardiographic screening and registration in a risk evaluation and mitigation strategy program to monitor HF symptoms and changes in ejection fraction and LVOTO. In patients who develop systolic dysfunction (LVEF ≤50%), interruption and continuation at a lower dose (if EF improves) or discontinuation (persistent EF ≤50%) of CMIs is a class I recommendation in the 2024 AHA/ACC HCM guidelines.46
The management of symptomatic non-obstructive HCM with preserved EF is challenging. β-blockers and non-dihydropyridine calcium channel blockers are recommended for patients with exertional angina or dyspnea. The addition of oral diuretics can be considered in cases of persistent exertional dyspnea. Based on the findings of the VANISH trial, the 2024 HCM AHA/ACC guidelines now include valsartan as a class 2b recommendation in younger patients (≤45 years) with non-obstructive HCM.46,47 There is also new evidence to suggest that sodium–glucose cotransporter 2 inhibitors may improve diastolic function and functional capacity in patients with non-obstructive HCM and preserved LV function.56 For those who develop systolic dysfunction, standard guideline-directed medical therapy is indicated. Advanced therapies, including cardiac transplantation, should be considered for individuals with HCM who develop severe, persistent heart failure symptoms due to severe restrictive LV dysfunction or severe LV systolic dysfunction.
Anticoagulation with a direct anticoagulant first line or vitamin K antagonist second line is recommended in all patients with HCM and clinical AF or subclinical AF ≥ 24 hours duration irrespective of CHA2DS2-VASc score, given their elevated stroke risk.46 Rare paroxysms of AF can be managed by rate control alone, but antiarrhythmics and catheter ablation can be considered for those with persistent arrhythmia.46
Procedural Management
SRT with either surgical myectomy or alcohol septal ablation is generally reserved for patients with severe symptoms (NYHA class III–IV) that are refractory to medical treatment.20 Surgical myectomy has been considered the gold standard for SRT.57
However, the widespread use of surgical myectomy was restricted by problems, such as limited access to centers with the necessary surgical expertise for the procedure and the increased risk associated with it, particularly in older patients or those with comorbidities. This led to the adoption of percutaneous alcohol septal ablation (ASA) as an effective alternative to surgical myectomy in selected patients. The effectiveness of this novel non-surgical, catheter-based technique was first demonstrated by Dr Ulrich Sigwart in 1994 at Royal Brompton Hospital, London, UK, in three patients with obstructive HCM.58 It involves the selective infusion of absolute alcohol into the first or largest septal branch of the left anterior descending artery to produce a localized infarct in the septum with consequent remodeling of the LV outflow tract. Based on current data, myectomy is superior to ASA in achieving optimal hemodynamic and symptomatic benefit, resulting in an improved quality of life.18
As per the latest 2020 ACC/AHA guidelines, surgical myectomy is recommended for patients with symptomatic obstructive HCM refractory to medical treatment and associated cardiac disease requiring surgical intervention.20 Surgical myectomy performed for symptom improvement and reduced LV outflow obstruction is associated with a reduction in mortality risk and long-term survival comparable with the general population.59 For patients where surgery is contraindicated or high risk, particularly in older people or those with comorbidities, ASA is the preferred option when performed in experienced centers.20 However, there is higher incidence of heart block requiring pacemaker implantation after ASA compared to surgery.60
Overall, the risks of SRT depend on proper patient selection and operator experience, and the current mortality associated with myectomy at dedicated HCM centers is low (≤0.6%), contradictory to the earlier view that surgery is a high-risk option for those with obstructive HCM.61
SRT may be combined with mitral valve replacement or leaflet plication in cases of obstructive HCM and severe MR due to SAM. Transcatheter edge-to-edge repair, such as with the MitraClip device (Abbott), may also be considered in patients with severe MR related to SAM, or in those with primary MR due to MV leaflet defects and non-obstructive HCM with severe symptoms that are refractory to medical management and who are at prohibitive surgical risk.62
Genetic Testing
A majority of cases of HCM are inherited in an autosomal dominant pattern.63 Mutations in a single gene encoding various components of the cardiac sarcomere, including the thick myofilament proteins (45%), thin myofilament proteins (5%), and Z-disc proteins (1%), are responsible for the development of cardiac hypertrophy.3
MYH7 and MYBPC3, which encode for myosin heavy chain and myosin binding protein C, respectively, are the two most prevalent genes implicated in familial HCM.64 Other common mutations include a mutation in PRKAG2, responsible for encoding the γ subunit of protein kinase A, which is associated with the distinct phenotype of HCM and Wolff-Parkinson-White syndrome.65 TNNT2 mutations, which encode for cardiac troponin T, have been associated with no or mild LVH, but a high risk of SCD.66
Initial genetic testing is usually performed in the index (proband) case. If a disease-causing variant has been identified, targeted testing of first-degree relatives can be considered.20 Nonetheless, if genetic testing is negative, echocardiogram and EKG screening is recommended for all first-degree family members. Current guidelines recommend screening at puberty, then every 1–2 years for children and every 3–5 years for adults.20 Routine genetic testing is not recommended for assessing prognosis or risk stratification in HCM, because the presence of sarcomere mutations does not reliably predict the disease’s clinical course. Regardless of the results of genetic testing, genetic counseling is essential for all patients and families affected by HCM, including before conception, to ensure informed decision-making.20
Sudden Cardiac Death and ICD Evaluation
The annual incidence of SCD in adults with HCM is 1%.67 The risk of SCD in a patient with HCM is determined based on their personal and family history and findings on noninvasive imaging, such as echocardiography, ambulatory electrocardiographic monitoring, and cardiac MRI. The key factors used to evaluate risk include a previous SCD event, ventricular fibrillation or sustained ventricular tachycardia, a family history of SCD in a first-degree relative aged ≤50 years, one or more unexplained syncope events occurring within 6 months of evaluation, massive LVH (>30 mm in any segment), diffuse LGE (≥15% of LV mass), LV apical aneurysm and reduced LVEF (≤50%).20 Maximal wall thickness and LV apical aneurysm may not be accurately characterized on echocardiography. In such instances, contrast-enhanced cardiac MRI is useful to more accurately evaluate these criteria for appropriate SCD risk stratification.68
The AHA/ACC guidelines recommend assessing SCD risk at the initial visit and then every 1–2 years in patients with HCM. Patients with HCM may be eligible for primary prevention ICD therapy in the presence of one or more SCD risk factors. Those who have experienced a cardiac arrest or significant episode of ventricular tachycardia or ventricular fibrillation may be considered for secondary prevention ICD therapy.20 Shared decision-making with the patient prior to ICD therapy is paramount, and this involves discussing the risk of SCD, the potential benefits of ICD implantation, and addressing the long-term complications associated with device therapy.
Exercise
The 2024 AHA/ACC HCM guidelines recommend mild to moderate intensity recreational physical activity in patients with HCM. For patients with minimal or no symptoms, participation in rigorous recreational activity or competitive sports can be considered following an annual comprehensive evaluation and discussion of the potential benefits and risks with an HCM expert. This marks a change from the previous 2020 AHA/ACC HCM guidelines, which universally restricted vigorous exercise for all patients with HCM, given recent data from LIVE-HCM, a multicenter prospective cohort study of 1,660 patients with HCM or who are genotype positive without LVH, revealed that vigorous exercise was not associated with a higher rate of death or life-threatening arrhythmias when compared to moderate-intensity or sedentary activity levels.69
Conclusion
HCM is a complex clinical condition that requires a detailed evaluation to confirm the diagnosis, screen for genetic predisposition and risk stratify for SCD. There are specific historical, physical examination and imaging findings that ensure prompt recognition and distinction from potential mimickers, such as HFpEF or LVH, thus enabling accurate diagnosis and treatment.
Medical therapy should be considered with β-blockers and calcium channel blockers as first-line agents, but CMIs have been proven effective as a novel therapy for the treatment of symptomatic obstructive HCM and may delay the need for septal reduction therapy.
The wide variety of phenotypes and clinical presentations can complicate the diagnosis of HCM but, with thorough and focused diagnostic evaluation, patients can be identified and appropriately managed, minimizing their morbidity and mortality.