A Hispanic man in his 50s with a history of obstructive sleep apnea, hypertension, hyperlipidemia, and HIV (on medications) was hospitalized for chest pain.
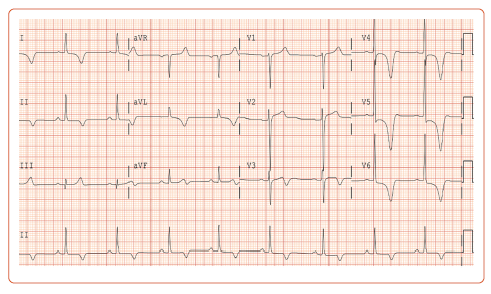
His blood pressure on presentation was 181/99 mmHg and heart rate was normal and in sinus rhythm. The physical examination was remarkable for a systolic ejection murmur at the left lower sternal border with no appreciable change with handgrip or Valsalva maneuver. An ECG was obtained, which showed deep T wave inversions in the lateral precordial and limb leads (Figure 1). Cardiac biomarkers were negative.
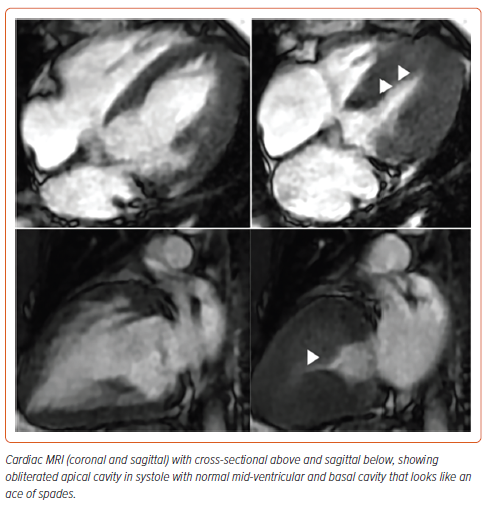
The patient underwent gadolinium-enhanced cardiac MRI which showed a thickened septum and true left ventricle apex with no aneurysms in coronal and cross-sectional views (Figure 2). Coronary angiography was negative for obstructive epicardial coronary artery disease. Thus, the patient was diagnosed with apical hypertrophic cardiomyopathy. He was discharged on metoprolol and valsartan.
The apical variant of hypertrophic cardiomyopathy (HCM) is less common than classic (septal) HCM, and is rare in non-Asians.1,2 Sarcomere mutations are detected less frequently in apical than in classic HCM. Patients have a higher prevalence of AF but are less likely to have left ventricular outflow tract obstruction. They also have more apical aneurysms. As with classic HCM, β-blockers are the first-line medical therapy.
There is no role for septal reduction therapies such as alcohol septal ablation or surgical myectomy in apical HCM due to the absence of overt septal hypertrophy in this variant. Finally, no models exist to predict mortality and requirement for ICD implantation in this specific variant of HCM.